UNIVERSITY 3F ALBERTA Hvestigation of Stereoselection By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chiral Proton Catalysis: Design and Development of Enantioselective Aza-Henry and Diels-Alder Reactions
CHIRAL PROTON CATALYSIS: DESIGN AND DEVELOPMENT OF ENANTIOSELECTIVE AZA-HENRY AND DIELS-ALDER REACTIONS Ryan A. Yoder Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Doctor of Philosophy in the Department of Chemistry, Indiana University June 2008 Accepted by the Graduate Faculty, Indiana University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Doctoral Committee _____________________________ Jeffrey N. Johnston, Ph.D. _____________________________ Daniel J. Mindiola, Ph.D. _____________________________ David R. Williams, Ph.D. _____________________________ Jeffrey Zaleski, Ph.D. April 24, 2008 ii © 2008 Ryan A. Yoder ALL RIGHTS RESERVED iii DEDICATION This work is dedicated to my parents, David and Doreen Yoder, and my sister, LeeAnna Loudermilk. Their unwavering love and support provided the inspiration for me to pursue my dreams. The sacrifices they have made and the strength they have shown continue to motivate me to be a better person each and every day. Thank you mom, dad, and little sis for being the rocks that I can lean on and the foundation that allowed me to find the happiness I have today. Without you, none of this would have been possible. iv ACKNOWLEDGEMENTS First and foremost I want to thank my research advisor, Professor Jeffrey N. Johnston. I am incredibly grateful for his mentoring and guidance throughout my time in the Johnston group. He has instilled in me a solid foundation in the fundamentals of organic chemistry and at the same time has taught me how to apply innovative and creative solutions to complex problems. -

Hydrogenation of Styrene Oxide to 2-Phenylethanol Over Nanocrystalline Ni Prepared by Ethylene Glycol Reduction Method
Hindawi Publishing Corporation International Journal of Chemical Engineering Volume 2014, Article ID 406939, 6 pages http://dx.doi.org/10.1155/2014/406939 Research Article Hydrogenation of Styrene Oxide to 2-Phenylethanol over Nanocrystalline Ni Prepared by Ethylene Glycol Reduction Method Sunil K. Kanojiya,1 G. Shukla,1 S. Sharma,1 R. Dwivedi,2 P. Sharma,2 R. Prasad,2 M. Satalkar,3 and S. N. Kane3 1 IPCA Laboratories Ltd., Indore 452003, India 2 School of Chemical Sciences, DA University, Indore 452001, India 3 Magnetic Materials Laboratory, School of Physics, DA University, Indore 452001, India Correspondence should be addressed to R. Prasad; [email protected] Received 14 February 2014; Accepted 20 May 2014; Published 17 July 2014 Academic Editor: Moshe Sheintuch Copyright © 2014 Sunil K. Kanojiya et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Nanocrystalline nickel prepared by glycol reduction method and characterized by XRD and magnetic measurements has been used as a catalyst for hydrogenation of styrene oxide to 2-phenylethanol. Effect of process variables such as particle size of the catalyst, temperature, and pressure have been optimized to achieve a maximum conversion of 98% of styrene oxide with 99% selectivity towards 2-phenylethanol. The structure of the transition state has been computed employing density functional theory −1 and using Gaussian 09 suite. The enthalpy of reaction (Δ) and activation energy ()arecalculatedtobe85.3kcal⋅mol and −1 123.03 kcal⋅mol , respectively. A tentative mechanism for the reaction is proposed according to which atomized hydrogen and styrene oxide react together over the catalyst surface to produce 2-phenylethanol. -
![Chiral Phosphorus Containing Calix[4]Arenes for Asymmetric Catalysis Andrii Karpus](https://docslib.b-cdn.net/cover/6317/chiral-phosphorus-containing-calix-4-arenes-for-asymmetric-catalysis-andrii-karpus-936317.webp)
Chiral Phosphorus Containing Calix[4]Arenes for Asymmetric Catalysis Andrii Karpus
Chiral phosphorus containing calix[4]arenes for asymmetric catalysis Andrii Karpus To cite this version: Andrii Karpus. Chiral phosphorus containing calix[4]arenes for asymmetric catalysis. Catalysis. Uni- versité Paul Sabatier - Toulouse III, 2017. English. NNT : 2017TOU30045. tel-01811136 HAL Id: tel-01811136 https://tel.archives-ouvertes.fr/tel-01811136 Submitted on 8 Jun 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 5)µ4& &OWVFEFMPCUFOUJPOEV %0$503"5%&-6/*7&34*5²%&506-064& %ÏMJWSÏQBS Université Toulouse 3 Paul Sabatier (UT3 Paul Sabatier) Cotutelle internationale avec Université nationale Taras-Chevtchenko de Kiev 1SÏTFOUÏFFUTPVUFOVFQBS KARPUS Andrii le mardi 24 janvier 2017 5JUSF Calix[4]arènes chiraux contenant des groupes phosphine comme ligands pour la catalyse ²DPMF EPDUPSBMF et discipline ou spécialité ED SDM : Chimie organométallique de coordination - CO 043 6OJUÏEFSFDIFSDIF CNRS - UPR 8241 - Laboratoire de Chimie de Coordination (LCC) %JSFDUFVSUSJDF T EFʾÒTF Dr. MANOURY Eric Pr. VOITENKO Zoia Jury : Dr. MANOURY Eric, Directeur de thèse Pr. VOITENKO Zoia, Co-directrice de thèse Dr. SEMERIL David, Rapporteur Pr. KOROTKIKH Nikolay, Rapporteur Pr. GENISSON YVES, Examinateur Dr. SMOLII Oleg, Examinateur Université Toulouse III Paul Sabatier (UT3 Paul Sabatier) Andrii O. -

Green Chemistry : Greener Alternatives to Synthetic Organic Transformations
Green Chemistry Greener Alternatives to Synthetic Organic Transformations V.K. Ahluwalia Alpha Science International Ltd. Oxford, U.K. Contents Preface v Parti 1. Introduction 1.1-1.10 1.1 Principles of Green Chemistry 1.1 1.2 How to Plan a Green Synthesis 1.2 Part II Green Alternatives to Synthesis Organic Transformations 2. Aqueous Phase Transformations 2.1-2.48 2.1 p-Acetylaminophenol (Tylenol) 2.1 2.2 3-Aminopyridine 2.2 2.2a Anthranilic Acid 2.3 2.3 Benzilic Acid 2.3 2.4 Benzoin 2.5 2.5 Benzotriazole 2.6 2.6 2-Benzoyl-3,5-dimethylbenzofuran 2.7 2.7 n-Butyl Bromide 2.8 2.8 Tert.Butylchloride 2.8 2.9 Chalcone (Benzalacetophenone) 2.9 2.10 Cycloheptanone 2.10 2.11 2,3-Dihydroxy Anisole (Pyrigallol Monomethyl-ether) 2.12 2.12 2,4-Dihydroxybenzoic acid (P-resorcylic Acid) 2.13 2.13 3,4-Dimethoxyphenol 2.14 2.14 2,3-Dimethyl-l-phenylpyrazol-5-one 2.15 2.15 3, 5-Dimethylpyrazole 2.16 2.16 5,5-Diphenylhydantoin 2.17 2.17 Endo-cis-1,4-endoxo-A5-cyclohexene-2,3-dicarboxylic Acid 2.18 2.18 p-Ethoxyacetanilide (Phenacetin) 2.19 2.19 6-Ethoxycarbonyl-3,5-diphenyI-2-cyclohexenone 2.20 2.20 HeteroDiels-AlderAdduct 2.22 2.21 Hippuric Acid (Benzoyl Glycine) 2.22 Viii Contents 2.22 Hydantion 2.23 2.25 3-Hydroxy-3-phenyl-2-methylene Proponamide 2.24 2.24 Iodoform 2.25 2.25 Inodole 2.26 2.26 3-(p-Methoxyphenyl)-2H-1,4-Benzoxazine 2.27 2.27 3-Methylcyclopent-2-enone 2.27 2.28 2-(2'-Methlindol-3yl)-1,4-benzoquinone 2.28 2.29 2-methyl-2-(3-oxobutyl)-1,3-cyclopentanedione 2.29 2.30 P-Naphthyl Acetate 2.30 2.31 (5-Naphthyl Methyl Ether (Nerolin) 2.31 2.32 -

Inhibition of Growth, Synthesis, and Permeability in Neurospora Crassa by Phenethyl Alcohol
JOURNAL OF BACTERIOLOGY, JUly, 1965 Vol. 90, No. I Copyright © 1965 American Society for Microbiology Printed in U.S.A. Inhibition of Growth, Synthesis, and Permeability in Neurospora crassa by Phenethyl Alcohol GABRIEL LESTER Department of Biology, Reed College, Portland, Oregon Received for publication 22 January 1965 ABSTRACT LESTER, GABRIEL (Reed College, Portland, Ore.). Inhibition of growth, synthesis, and permeability in Neurospora crassa by phenethyl alcohol. J. Bacteriol. 90: 29-37. 1965.- Inhibition of the growth of Neurospora crassa in still culture was detected at 0.05% and was complete at a level of 0.2% phenethyl alcohol (PEA). Benzyl alcohol was less in- hibitory, and 3-phenyl-1-propanol and phenol were more inhibitory, than PEA; benzyl- amine and phenethylamine were less inhibitory than the analogous hydroxylated com- pounds. Inhibition by PEA was not reversed by synthetic mixtures of purines and pyrimidines or vitamins, or by casein digests, yeast extract, or nutrient broth. The germination of conidia was inhibited by PEA, but after an exposure of 8.5 hr no loss of viability was observed. The addition of PEA to growing shake cultures caused a simul- taneous inhibition of growth and of the syntheses of ribonucleic and deoxyribonucleic acids and protein; the relationships of these compounds to mycelial dry weight and to one another were constant in growing mycelia, and PEA did not significantly affect these relationships. PEA partially inhibited the uptake of glucose, but severely re- stricted the accumulation of L-leucine, L-tryptophan, or a-aminoisobutyric acid in germinated conidia. The efflux of a-aminoisobutyric acid from germinated conidia was somewhat enhanced by PEA, but this effect was not so pronounced as the (complete) inhibition of a-aminoisobutyric acid accumulation by PEA. -

United States Patent (19) 11 4,267,375 Maasbol Et Al
United States Patent (19) 11 4,267,375 Maasbol et al. 45 May 12, 1981 54 PREPARATION OF THIOETHERS OTHER PUBLICATIONS 75) Inventors: Alfred G. Maasbol, Hamburg, Fed. I. Ruderman et al., J. Amer. Chem. Soc., 71, pp. Rep. of Germany; Lothar G. Dulog, 2264-2265, (1949). St. Martens Latem, Belgium Morrisson and Boyd, Organic Chemistry, 2nd edition, (1967), pp. 29–30. 73) Assignee: s.a. Texaco Belgium in.v., Brussels, T. Todsen et al., J. Amer. Chem. Soc., 72, pp. Belgium 4000-4002, (1950). Berichte Deutsch. Chemie, vol. 1, pp. 587-591, (1935), (21) Appl. No.: 945,273 Berlin. 22 Filed: Sep. 25, 1978 D. Gregg et al., J. Org. Chem., pp. 246-252, (1950). M. Malinovskii, Epoxides and Their Derivatives, pp. Related U.S. Application Data 131-136, (1965), Jerusalem, Daniel Davey & Co. Primary Examiner-Glennon H. Hollrah 63 Continuation of Ser. No. 703,045, Jul. 6, 1976, aban Assistant Examiner-M. C. Eakin doned. Attorney, Agent, or Firm-Carl G. Ries; Robert A. 30 Foreign Application Priority Data Kulason; Carl G. Seutter Nov. 19, 1975 GB United Kingdom ............... 47582/75 57 ABSTRACT .. 51 Int. Cl. ............................................ CO7C 149/30 Thioethers may be prepared by reacting a thiol, such as thiophenol, with an alcohol (having electron donor 52 U.S. C. ......................................... 568/57; 568/58 groups in the alpha or beta position to its hydroxyl 58 Field of Search ........................ 260/609 E, 609 R group) such as phenyl-1-hydroxy-phenethylsulfide. Re 56) References Cited action is carried out in the presence of a Lewis Acid U.S. PATENT DOCUMENTS metal halide, typically zinc chloride. -
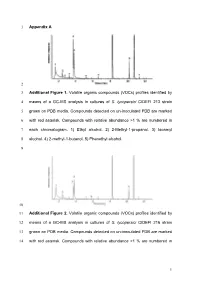
Appendix a 1 2 Additional Figure 1. Volatile Organic Compounds (Vocs
1 Appendix A 2 3 Additional Figure 1. Volatile organic compounds (VOCs) profiles identified by 4 means of a GC-MS analysis in cultures of S. lycopersici CIDEFI 213 strain 5 grown on PDB media. Compounds detected on un-inoculated PDB are marked 6 with red asterisk. Compounds with relative abundance >1 % are numbered in 7 each chromatogram. 1) Ethyl alcohol. 2) 2-Methyl-1-propanol. 3) Isoamyl 8 alcohol. 4) 2-methyl-1-butanol. 5) Phenethyl alcohol. 9 10 11 Additional Figure 2. Volatile organic compounds (VOCs) profiles identified by 12 means of a GC-MS analysis in cultures of S. lycopersici CIDEFI 216 strain 13 grown on PDB media. Compounds detected on un-inoculated PDB are marked 14 with red asterisk. Compounds with relative abundance >1 % are numbered in 1 15 each chromatogram. 1) Ethyl alcohol. 2) 2-Methyl-1-propanol. 3) Isoamyl 16 alcohol. 4) 2-methyl-1-butanol. 5) Phenethyl alcohol. 6) Furfuryl alcohol. 17 18 19 Additional Figure 3. Volatile organic compounds (VOCs) profiles identified by 20 means of a GC-MS analysis in cultures of F. fulva CIDEFI 300 strain grown on 21 PDB media. Compounds detected on un-inoculated PDB are marked with red 22 asterisk. Compounds with relative abundance >1 % are numbered in each 23 chromatogram. 6) Furfuryl alcohol. 7) Acetone. 8) Methyl trimethylacetate. 9) 24 Isoamyl alcohol. 10) 1-Octene. 11) 3-Hexanone, 4-methyl-. 12) Styrene. 13) 3- 25 Octanone. 14) Hexanoic acid, 2 ethyl-, methyl ester. 15) 2-Nonanone. 16) 26 Phenethyl alcohol. 17) No identified Nist05. 27 2 28 29 Additional Figure 4. -

Regioselective and Stereodivergent Synthesis of Enantiomerically Pure Vic-Diamines from Chiral Β-Amino Alcohols with 2-Pyridyl and 0 † 6-(2,2 -Bipyridyl) Moieties
molecules Article Regioselective and Stereodivergent Synthesis of Enantiomerically Pure Vic-Diamines from Chiral β-Amino Alcohols with 2-Pyridyl and 0 y 6-(2,2 -Bipyridyl) Moieties Marzena Wosi ´nska-Hrydczuk,Przemysław J. Boraty ´nski and Jacek Skar˙zewski* Chair of Organic and Medicinal Chemistry, Faculty of Chemistry, Wrocław University of Science and Technology, Wyb. Wyspia´nskiego27, 50-370 Wrocław, Poland; [email protected] (M.W.-H.); [email protected] (P.J.B.) * Correspondence: [email protected]; Tel.: +48-71-320-2464 This paper is dedicated to Professor Grzegorz Mlosto´non the occasion of his 70-th birthday. y Academic Editors: Zbigniew Czarnocki and Joanna Szawkało Received: 11 January 2020; Accepted: 6 February 2020; Published: 7 February 2020 Abstract: In this report, we describe the synthetic elaboration of the easily available enantiomerically pure β-amino alcohols. Attempted direct substitution of the hydroxyl group by azido-functionality in the Mitsunobu reaction with hydrazoic acid was inefficient or led to a diastereomeric mixture. These outcomes resulted from the participation of aziridines. Intentionally performed internal Mitsunobu reaction of β-amino alcohols gave eight chiral aziridines in 45–82% yield. The structural and configuration identity of products was confirmed by NMR data compared to the DFT calculated GIAO values. For 1,2,3-trisubstituted aziridines slow configurational inversion at the endocyclic nitrogen atom was observed by NMR at room temperature. Moreover, when aziridine was titrated with Zn(OAc)2 under NMR control, only one of two N-epimers directly participated in complexation. The aziridines underwent ring opening with HN3 to form the corresponding azido amines as single regio- and diastereomers in 90–97% yield. -

Chemicals That Form Explosive Levels of Peroxides Without Concentration (Safe Storage Time After Opening - 3 Months) Chemical CAS # Synonym State Ref
Group A- Chemicals that form explosive levels of peroxides without concentration (Safe storage time after opening - 3 months) Chemical CAS # Synonym State Ref. 000106- Butadiene(1,3) 1,3-Butadiene gas 4 99-0 000126- 2-Chloro-1,3- Chloroprene (1,3) liquid 4 99-8 butadiene 000821- Divinyl acetylene 1,5-Hexadien- 3-yne liquid 5 08-9 000108- Isopropyl ether liquid 5 20-3 000116- Tetrafluoroethylene gas 4 14-3 000109- Vinyl ether Divinyl ether liquid 5 93-3 000075- 1,1- Vinylidene chloride liquid 5 35-4 Dichloroethylene Group B-Chemicals that form explosive levels of peroxides on concentration (Safe storage time after opening - 12 months) Chemical CAS # Synonym State Ref. 000105- Acetal liquid 5 57-7 000075- Acetaldehyde liquid 4 07-0 000100- Benzyl alcohol liquid 4 51-6 000078- 2-Butanol liquid 4 92-2 000108- Cyclohexanol liquid 4 93-0 000110- Cyclohexene liquid 5 83-8 000822- 2-Cyclohexen-1-ol liquid 4 67-3 000142- Cyclopentene liquid 5 29-0 000091- Decahydronaphthalene liquid 4 17-8 000460- Diacetylene gas 5 12-8 000077- Dicyclopentadiene liquid 5 73-6 Diethylene glycol 000111- Diglyme liquid 5 dimethyl ether 96-6 000123- Dioxane 1,4-Dioxane liquid 5 91-1 Ethylene glycol 000110- Glyme liquid 5 dimethyl ether 71-4 000060- Ethyl ether Diethyl ether liquid 5 29-7 000110- Furan liquid 5 00-9 000589- 4-Heptanol liquid 4 55-9 000626- 2-Hexanol liquid 4 93-7 000098- Isopropyl benzene Cumene liquid 5 82-8 000074- Methyl acetylene Propyne gas 5 99-7 000123- 3-Methyl-1-butanol Isoamyl alcohol liquid 4 51-3 000096- Methyl cyclopentane liquid 5 37-7 -

Synthesis of Peptide and Protein Thioesters Through an N→S Acyl Shift
Native Chemical Thioesterification: Synthesis of Peptide and Protein Thioesters through an N→S Acyl Shift Jaskiranjit Kang A thesis submitted in partial fulfilment of the requirements for the degree award of: Doctor of Philosophy University College London Department of Chemistry 2010 Native Chemical Thioesterification: Synthesis of Peptide and Protein Thioesters through an N→S Acyl Shift Declaration I, Jaskiranjit Kang, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 Native Chemical Thioesterification: Synthesis of Peptide and Protein Thioesters through an N→S Acyl Shift Abstract The total chemical synthesis of a protein provides atomic-level control over its covalent structure, however polypeptides prepared by solid phase peptide synthesis are limited to approximately fifty amino acid residues. This limitation has been overcome by 'Native Chemical Ligation‘, which involves amide bond formation between two unprotected polypeptides: a peptide with a C-terminal thioester and an N-terminal cysteinyl peptide. Synthesis of the required peptide thioester is difficult, particularly by Fmoc-chemistry. During our studies towards the semisynthesis of erythropoietin, we discovered reaction conditions that reversed Native Chemical Ligation and generated peptide and protein thioesters through an N→S acyl transfer. O HS H3N O O O + H3O RSH N S SR H O A peptide with both a Gly-Cys and an Ala-Cys-Pro-glycolate ester sequence was selectively thioesterified between the Gly-Cys sequence upon microwave-heating at 80 °C with 30 % v/v 3-mercaptopropionic acid (MPA), to afford the peptide-Gly-MPA thioester (84 % yield). -

Asymmetric Total Synthesis of Congeners of Hydramycin, an Anthraquinone-Type Antitumor Agent
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 12-2011 Asymmetric Total Synthesis of Congeners of Hydramycin, an Anthraquinone-Type Antitumor Agent Costyl Ngnouomeuchi Njiojob [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Heterocyclic Compounds Commons, Organic Chemicals Commons, and the Polycyclic Compounds Commons Recommended Citation Njiojob, Costyl Ngnouomeuchi, "Asymmetric Total Synthesis of Congeners of Hydramycin, an Anthraquinone-Type Antitumor Agent. " PhD diss., University of Tennessee, 2011. https://trace.tennessee.edu/utk_graddiss/1210 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Costyl Ngnouomeuchi Njiojob entitled "Asymmetric Total Synthesis of Congeners of Hydramycin, an Anthraquinone-Type Antitumor Agent." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Chemistry. David C. Baker, Major Professor We have read this dissertation and recommend its acceptance: Shawn Campagna, Ben Xue, Elizabeth Howell Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Asymmetric Total Synthesis of Congeners of Hydramycin, an Anthraquinone-Type Antitumor Agent A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Costyl Ngnouomeuchi Njiojob December 2011 DEDICATION This dissertation is dedicated to my Parents Francois Ngnouomeuchi and Lydie T. -

CHM205 Chemicals by Experiment Tuesday, November 17, 2015 3:14:15 PM Experiment Title Chemical Name Concentration Acetaminophen Synthesis Acetic Anhydride Liquid
CHM205 Chemicals by Experiment Tuesday, November 17, 2015 3:14:15 PM Experiment Title Chemical Name Concentration Acetaminophen Synthesis Acetic anhydride liquid Acetaminophen Synthesis p-aminophenol solid Alcohols to Alkyl chlorides 2-pentanol liquid Alcohols to Alkyl chlorides Hydrochloric acid 12 M Alcohols to Alkyl chlorides Sodium carbonate solid Alcohols to Alkyl chlorides Hydrobromic acid 48% w/v Alcohols to Alkyl chlorides Sodium sulfate anhydrous solid Alcohols to Alkyl chlorides sec-phenethyl alcohol liquid Alcohols to Alkyl chlorides Benzyl alcohol liquid Alcohols to Alkyl chlorides t-butanol liquid Alcohols to Alkyl chlorides 1-pentanol liquid Alcohols to Alkyl chlorides Sodium carbonate 10% w/v Diels Alder Reaction 2,3-dimethyl-1,3-butadiene liquid Diels Alder Reaction Maleic anhydride solid Diels Alder Reaction Ethanol 95% Liquid Diels Alder Reaction Hexane liquid Diels Alder Reaction Cyclohexane liquid Diels Alder Reaction Calcium chloride solid Esterification methanol liquid Esterification Sodium carbonate 10% w/v Esterification 1-propanol liquid Esterification 1-butanol liquid Esterification trans-cinnamic acid solid Esterification Isoamyl alcohol liquid Esterification Isopropyl alcohol liquid Esterification Benzyl alcohol liquid Esterification Sulfuric acid conc. 18 M Esterification 1-pentanol liquid Esterification Isobutyl alcohol liquid Esterification Ethanol 95% liquid Page 1 of 3 Experiment Title Chemical Name Concentration Extraction of Beta Carotene Cyclohexane liquid Extraction of Beta Carotene Beta carotene UV