Mechanisms of Inflammation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rest Lawn Memorial Park Burial Book 1
REST LAWN MEMORIAL PARK BURIAL BOOK Surname FIRST NAME BIRTH-DATE DEATH or GARD SEC BLK PLOT BURIAL DATE EN Abbey Gaither William 1939 25-Mar-1963 BET 273 A 1 Abbey Geneva Hope 10-Oct-1919 18-Oct-1942 BET 273 A 2 Ackerby Seth 08/27/1878 30-Mar-1953 HOP 14 2 Adams Bertha V. 11-Jan-1909 13-Feb-2001 ARB 136 B 10 Adams Carsen 24-Dec-1918 30-Oct-1971 BET 229 B 9 Adams Eleanor 2-Nov-1937 15-Dec-2000 BET 216 B 3 Adams Elmer C. 09/18/1888 30-Aug-1981 CED 364 3 Adams Elmer Floyd 27-Jan-1917 20-Jul-1960 CED 364 6 Adams Gary Pearl 28-Apr-1941 23-Oct-1996 BET 229 B 8 Adams James Victor 12-Mar-1901 7-Mar-1974 ARB 136 B 9 Adams Lucille 4-Dec-1920 12-Jun-2013 BET 229 B 10 Adams Mabel A. 09/14/1888 28-Nov-1984 CED 364 4 Adams Marion A. 21-Apr-1928 14-Aug-2000 ARB 136 B 7 Agee Della E. 1874 15-Apr-1951 BET 249 A 2 Agee James R. 1868 15-Jul-1936 BET 249 A 1 Aho Beatrice Thompson 18-Jun-1914 28-May-1999 BET 185 B 2 Aho Toivo 9-Mar-1908 27-Feb-1983 BET 186 B 2 Akers Bertha M. 16-May-1909 9-Oct-1910 ARB 30 0 1 Akers Izorab 02/07/1863 05/08/1889 ARB 68 0 5 Akers Jabez H. 04/15/1827 30-Sep-1909 ARB 68 0 4 Akers Mabel Hoel 1879 28-Feb-1962 ARB 30 0 3 Akers Medley E. -

8Th International Conference on Isotopes and Expo
th 2 0 1 4 8th International Conference on Isotopes and Expo Preparing for Tomorrow Sponsored by the Accelerator Applications, Preliminary Program Biology & Medicine, and Isotope & Radiation Divisions of the American Nuclear Society www.8ici.org August 24-28, 2014 Hyatt Regency-Chicago Chicago, IL SPONSORS Accelerator Applications Division Biology and Medicine Division Isotopes and Radiation Division 2 2014 International Conference on Isotopes and Expo: Preliminary Program www.ans.org Table of Contents Plenary Programs and Speakers Sponsors 2 Meeting Officials 4-5 Meeting Information and Special Events 6 Plenary Programs and Speakers 7 Meeting Schedule 8-9 Monday Technical Sessions 10-13 Tuesday Technical Sessions 14-18 Wednesday Technical Sessions 19-23 Thursday Technical Sessions 23-24 2014 8TH ICI Registration Form 25 www.8ici.org 2014 International Conference on Isotopes and Expo: Preliminary Program 3 Meeting Officials Honorary Chair: General Chair: Assistant General Chair: Myung-Chul Lee Paul T. Dickman Nigel R. Stevenson President, WCI Argonne National Laboratory Clear Vascular, Inc. President, Korean Association for Radiation Application Technical Program Co-Chair: Technical Program Co-Chair: Finance Chair: Rolf Zeisler Stephen P. LaMont James T. Tanner National Institute of Standard Los Alamos National Laboratory U.S. Food and Drug Administration and Technology (retired) Publications Co-Chair: Publications Co-Chair: International Program Director: Sam Glover Bryan P. Bednarz Gulbarshyn Bozheyeva University of Cincinnati University of Wisconsin-Madison MELE Associates, Inc. Executive Advisory Board Executive Advisory Board: Executive Advisory Board: JongKyung Kim Meera Venkatesh Ron Cameron WCI, Secretary-General IAEA OECD-NEA President, KAERI Executive Advisory Board Member: President Elect, WCI International Coordinator WCI: Ilham Y. -

2006 Annual Report Board of Directors Kathryn R
Legal Aid Society of Middle Tennessee and the Cumberlands 2006 Annual Report Board of Directors Kathryn R. Edge, President N. Houston Parks, First Vice President Susan L. Kay, Second Vice President Richard K. Evans, Third Vice President Valerie Martin, Secretary John Andrew Goddard, Treasurer John Pellegrin, Past President Charles H. Warfield, Executive Committee Member at Large Clisby Barrow John T. Blankenship Richard M. Brooks Melanie T. Cagle Robert Allen Dickens Roberta Dobbins Trudy Edwards Daniel B. Eisenstein Craig Fickling Barbara Gooch Fannie J. Harris Amy T. Hollars G. Wilson Horde Lou Lavender Turner McCullough, Jr. James D. Petersen Teresa Poston Adrie Mae Rhodes Steve Rhodey Denice Scott Keith S. Smartt Gregory D. Smith Guilford F. Thorton, Jr. James L. Weatherly, Jr. Shelby York Nashville Pro Bono Program Board Mary Griffin, Chair Andrée Blumstein Daniel B. Eisenstein Richard Green Tonya Mitchem Grindon Susan L. Kay N. Sue Van Sant Palmer Jonathan E. Richardson Robyn L. Ryan Thor Y. Urness Mark H. Westlake Message From the President of the Board Dear Friends and Colleagues: Legal Aid Society provides a helping hand to those who need it most – low-income citizens who cannot afford professional legal help and have nowhere to turn. When they receive the help they need, when they learn that there is justice out there for everyone, then the entire community benefits. America was founded on the ideal that everyone should be treated equally under the law. And while it is still possible to find examples of justice best serving those who can afford to pay for it, Legal Aid proves that the ideals which are fundamental to a fair and just society do, indeed, exist in our nation today. -

1980 Surname
Surname Given Age Date Page Maiden Note Abbett Howard 91 6-Jan D-2 Abercrombie Levi Sr. 30-Mar E-15 Able Cora Ree 73 4-Dec C-8 Acevedo Marcelina P. 68 11-Dec D-2 Acton John Wesley 85 12-May D-1 Adam Michael Sr. 88 7-Mar B-3 Adam Millee 59 3-Mar B-6 Adam Sophie (Sister Ann 66 15-Dec B-7 Madeline) Adamczyk Josephine 82 4-May D-6 Adams Francis (Sheik) 78 14-Jul C-5 Adams Ruth Carol 41 17-Oct C-5 Adams William H. 63 21-Aug B-2 Ade Eleanor Anne 63 17-Jul C-14 Adelsperger James F. 78 22-Dec C-5 Adkins Otis C. 67 24-Jun C-1 Adler Florian F. 83 10-Aug D-2 Afflek Day Malo 18-Mar B-4 Agosto Gregorio 56 3-Dec E-2 Ahlborn Rudolph C. 81 10-Sep E-1 Ahlborn Walter W. 78 20-Jul D-1 Aikman Myrtle M. 74 26-Nov D-1 Albert Joseph (Larry) 51 11-Jan B-4 Albertson Russell A. 83 27-Jul C-7 Alderden Gertrude 76 7-Jul B-4 Aleman Sadie 21-Jan B-5 Alexander Edward L. 46 5-Aug C-3 Alexander Robert W. Jr. 65 2-Jan C-9 Alexander Sonja E. 67 11-Feb C-4 Alier Audra 65 30-Nov E-11 Brown Allen Clarence F. 57 9-May B-5 Allen Norman 79 7-Aug C-4 Picture included. Allen Rabe (Ray) 95 22-Dec C-5 Allen William J. -
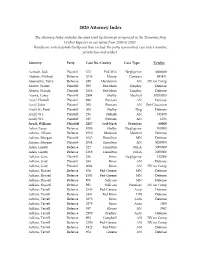
2020 Attorney Index
2020 Attorney Index The Attorney Index includes the cases tried by attorneys as reported in the Tennessee Jury Verdict Reporter in our issues from 2005 to 2020. Results are sorted alphabetically and then include the party represented, case index number, jurisdiction and verdict. Attorney Party Case No.-County Case Type Verdict Aamodt, Jodi Plaintiff 372 Fed-Win Negligence 8000000 Abelow, Michael Defense 2538 Maury Contract 995431 Abernathy, Terry Defense 240 Hardeman AN DV on Comp Abioto, Walter Plaintiff 508 Fed-Mem Employ Defense Abioto, Wanda Plaintiff 2618 Fed-Mem Employ Defense Acerra, Carey Plaintiff 2894 Shelby Medical 30035000 Acuff, Howell Plaintiff 920 Putnam AN Defense Acuff, John Plaintiff 292 Putnam AN Def-Causation Acuff, Jr., Fred Plaintiff 500 Shelby Dog Defense Acuff, W.I. Plaintiff 296 DeKalb AN 185100 Acuff, W.I. Plaintiff 285 Putnam AN 1233 Acuff, William Plaintiff 3257 Fed-Nash Premises 68000 Adair, Lacey Defense 1095 Shelby Negligence 100000 Adams, Allison Defense 2510 Madison Medical Defense Adams, Morgan Plaintiff 1625 Hamilton MN Defense Adams, Morgan Plaintiff 2068 Hamilton AN 9250000 Aden, Gareth Defense 522 Hamilton FELA 5000000 Aden, Gareth Defense 1368 Hamilton FELA 3000000 Adkins, Gary Plaintiff 216 Knox Negligence 132000 Adkins, Gary Plaintiff 838 Knox AN Defense Adkins, Gary Plaintiff 2084 Knox AN DV on Comp Adkins, Russell Defense 678 Fed-Greene MN Defense Adkins, Russell Defense 1503 Fed-Greene MN Defense Adkins, Russell Defense 976 Sullivan MN Defense Adkins, Russell Defense 981 Sullivan Premises DV -
The Bronte¨S in Context
Cambridge University Press 978-0-521-76186-4 - The Brontës in Context Edited by Marianne Thormählen Index More information Index Recipients of letters are not indexed in that capacity. Nor are names and titles in notes when indexed as occurring in the running text. Titles of works by authors other than the Bronte¨s are indexed (under their authors’ names) only when the primary reference in the running text is to the relevant work and not to its author. Ablow, Rachel, 203 Askew, Anne, 111 Abrams, M. H., 217 Athenaeum, The, 160, 165n, 181n, 274 Acton, Dr William, 329 Athena¨um, Das, 225 Adams, Abigail B., 120 Atlas, The, 161 Adams, J. F. A., 259n Audubon, John James, 144 adaptations of the Bronte¨ fiction, see screen Ornithological Autobiography, 250 versions of the Bronte¨ novels; sequels and Augustine, St, Confessions, 228 prequels to Bronte¨ fiction; stage versions Austen, Jane, 148, 306 of Bronte¨ novels Pride and Prejudice, 297, 301 advertising of books, 161 Austin, Linda, 203 Aesop’s Fables, 99, 144 Australia, deportation of criminals to, 230 agricultural revolution, 276, 281, 296 Author’s Printing and Publishing Assistant, The, agriculture, 276, 277–9 154, 155 Alcott, Louisa May, 201 Aykroyd, Tabitha, 76, 83–4, 98, 297 Alexander, Christine, 3, 60n, 89, 105n, 133n, Aylott and Jones, publishing firm, 154, 155, 160 157n, 171, 249n, 251, 259n, 270, 271; (ed.), 25n, 60n, 67n, 105n, 149n, 214n, 249n Babbage, Benjamin Herschel, 14, 25, 26n Allbutt, Sir Thomas Clifford, 51 Report, 336 Allen, David Elliston, 250 Bacon, Francis, The Advancement -
Schools Prepare for COVID Lawmakers Seek ‘Endangered’ Status for Local Protocols for Dealing with Virus Not Affected by Gov
Project1:Layout 1 6/10/2014 1:13 PM Page 1 Olympics: A look back at some top images of empathy /B1 TUESDAY T O DA Y C I T R U S C O U N T Y & n e x t m o r n i n g HIGH 91 Partly sunny; LOW scattered evening storms. 75 PAGE A4 www.chronicleonline.com AUGUST 10, 2021 Florida’s Best Community Newspaper Serving Florida’s Best Community $1 VOL. 126 ISSUE 307 NEWS BRIEFS Schools prepare for COVID Lawmakers seek ‘endangered’ status for Local protocols for dealing with virus not affected by Gov. DeSantis order manatees HANNAH signed into law House Bill Health and the Florida parents’ right under Flor- In addition, the order 241, the Parents’ Bill of Department of Education ida law to make health states the Florida Com- With Florida seeing a SACHEWICZ Staff writer Rights, which prevents to work together. care decisions for their missioner of Education record number of mana- any governmental institu- The goal of this partner- minor children; and pro- will pursue all legal tee deaths this year, two On July 30, Gov. Ron De- tion from making health ship is to ensure COVID- tect children with disabil- means available to ensure congressmen Monday Santis issued an execu- care decisions for minor 19 safety protocols that, ities or health conditions districts adhere to Flor- said they have introduced tive order, “Ensuring children. according to the executive who would be harmed by ida law, including with- legislation that would des- Parents’ Freedom to The governor’s execu- order, “do not violate Flo- certain protocols such as holding funding. -

Obituary "J" Index
Obituary "J" Index Copyright © 2004 - 2021 GRHS DISCLAIMER: GRHS cannot guarantee that should you purchase a copy of what you would expect to be an obituary from its obituary collection that you will receive an obituary per se. The obituary collection consists of such items as a) personal cards of information shared with GRHS by researchers, b) www.findagrave.com extractions, c) funeral home cards, d) newspaper death notices, and e) obituaries extracted from newspapers and other publications as well as funeral home web sites. Some obituaries are translations of obituaries published in German publications, although generally GRHS has copies of the German versions. These German versions would have to be ordered separately for they are kept in a separate file in the GRHS library. The list of names and dates contained herein is an alphabetical listing [by surname and given name] of the obituaries held at the Society's headquarters for the letter combination indicated. Each name is followed by the birth date in the first column and death date in the second. Dates may be extrapolated or provided from another source. Important note about UMLAUTS: Surnames in this index have been entered by our volunteers exactly as they appear in each obituary but the use of characters with umlauts in obits has been found to be inconsistant. For example the surname Büchele may be entered as Buchele or Bahmüller as Bahmueller. This is important because surnames with umlauted characters are placed in alphabetic order after regular characters so if you are just scrolling down this sorted list you may find the surname you are looking for in an unexpected place (i.e. -
Medical Lab Breach Leaks Data of 11.9M National Blood-Testing Center Has Columbia County Branch
A3 + PLUS >> May 35 matters, Opinion/4A BASEBALL LOCAL CHS to host City axes storm all-star games fee regulations See Page 5A Story below TUESDAY, JUNE 4, 2019 | YOUR COMMUNITY NEWSPAPER SINCE 1874 | $1.00 Lake City Reporter LAKECITYREPORTER.COM QUEST y g DIAGNOSTICS Heav dama e to interstate Medical lab breach leaks data of 11.9M National blood-testing center has Columbia County branch. From staff reports Quest Diagnostics, a national medical laboratory with a branch in Lake City, said Monday that the pri- vate information of nearly 12 million patients had been Social hacked. Security American numbers, Medical Collection financial Agency, a billing information collections ser- vice provider, told among data Quest an “unautho- stolen. rized user” gained access to AMCA’s system containing personal informa- tion AMCA received from companies including from Quest, according to a press release. AMCA provides billing collections services to Optum360, which in turn is a Quest contractor. Quest and Optum360 are working with forensic experts to investigate the breach, the release said. AMCA first notified Quest and Optum360 on May 14 of potential unau- BREACH continued on 2A COURTESY FDOT/FHP The shoulder of Interstate 75 bears the scars of a large piece of industrial equipment that fell off a semi passing beneath Woman faces on I-10 and tore holes in the roadway. The culprit (inset) may have been a wind turbine assembly, according to FHP. 2nd charge of Cargo comes dislodged child neglect By CARL MCKINNEY from semi, rips roadway [email protected] From staff reports the truck, the release said, The subject of an ongoing DUI and A piece of industrial equip- “then rolled down Interstate child neglect case has landed in trou- ment nearly the size of a 10, causing major damage to ble with the law again. -

Effects of A-Beta Immunotherapy and Omega-3 Fatty Acid Administration in Alzheimer's Transgenic Mice by Maren T. Jensen A
Effects of A-beta Immunotherapy and Omega-3 Fatty Acid Administration in Alzheimer’s Transgenic Mice by Maren T. Jensen A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Biology College of Arts and Sciences University of South Florida Major Professor: Gary Arendash, Ph.D. Huntington Potter, Ph.D. Sidney Pierce, Ph.D. Brian Livingston, Ph.D. Date of Approval: March 6, 2006 Keywords: Active Vaccination, Fish Oil, DHA, Cognitive Performance, Plaque Deposition © Copyright 2006, Maren T. Jensen Acknowledgements I thank Dr. David Morgan for providing the animals utilized in the Aβ immunotherapy study and Dr. Marcia Gordon for her involvement in the animal vaccinations and Aβ immunostaining. I would also like to thank Purina Mills Test Diet for providing the low omega-3, omega-3-deficient, high n-3 and high n-6 fatty acid diets and Barrow-Agee Laboratories for analyzing all four of the fatty acid diets. In addition, I would like to thank Dr. Norman Salem and Dr. Nahed Hussein for their determination of fatty acid levels from the frontal cortex. Table of Contents List of Tables ..................................................................................................................... iii List of Figures..................................................................................................................... v Introduction......................................................................................................................... 1 Alzheimer’s -

Poor Oversight Costs City Big Bucks Page 5
Vol. XXXV, Number 4 N November 1, 2013 Poor oversight costs city big bucks Page 5 www.PaloAltoOnline.com saving the Scientists, nonprofi t groups work to protect Palo Alto marshlands PAGE 33 Pulse 14 Spectrum 16 Transitions 20 Eating Out 23 Movies 25 Seniors 28 Puzzles 58 NArts Jérôme Bel: postmodern dance provocateur Page 21 NHome Fall fl oral wreaths from Hidden Villa Page 36 NSports Paly girls win CCS golf title Page 60 11/ 3 /13. Page 2ÊUÊ ÛiLiÀÊ£]ÊÓä£ÎÊUÊ*>ÊÌÊ7iiÞÊUÊÜÜÜ°*>Ì"i°V THANK YOU Jackie and Richard thank you for trusting us to help you achieve your Real Estate Success. 940 Monte Rose, Menlo Park* 678 College, Menlo Park 9 Atherton Oaks, Atherton* SOLD SOLD SOLD 96 Tuscaloosa, Atherton* 210 Montalvo, Emerald Hills 1003 Almanor, Menlo Park SOLD SOLD SOLD 1530 University, Palo Alto 98 Kilroy, Atherton* 1941 Deodara, Los Altos SOLD SOLD SOLD Call Jackie and Richard for Your Free Home Consultation Jackie Richard 650-855-9700 650-566-8033 [email protected] [email protected] BRE # 01092400 BRE # 01413607 www.schoelerman.com *represented the buyer ÜÜÜ°*>Ì"i°VÊUÊ*>ÊÌÊ7iiÞÊUÊ ÛiLiÀÊ£]ÊÓä£ÎÊU Page 3 www.deleonrealty.com Page 4ÊUÊ ÛiLiÀÊ£]ÊÓä£ÎÊUÊ*>ÊÌÊ7iiÞÊUÊÜÜÜ°*>Ì"i°V Daylight Saving Time is ending Set your clocks back one hour at 2 a.m. this Sunday. UpfrontLocal news, information and analysis City management lapse may have cost $281,000 New City Auditor report: Lack of oversight increased documents and adequately monitor of fraud, waste and abuse.” evaluate or renew the City’s con- risk of ‘fraud, waste and abuse’ the work being done. -

Economics of Radioisotope Production and Sustainability 1 Ron Cameron (OECD Nuclear Energy Agency), Invited
8th International Conference on Isotopes 2014 (8ICI) Chicago, Illinois, USA 24 – 28 August 2014 ISBN: 978-1-5108-0236-0 Printed from e-media with permission by: Curran Associates, Inc. 57 Morehouse Lane Red Hook, NY 12571 Some format issues inherent in the e-media version may also appear in this print version. Copyright© (2014) by the American Nuclear Society All rights reserved. Printed by Curran Associates, Inc. (2015) For permission requests, please contact the American Nuclear Society at the address below. American Nuclear Society 555 North Kensington Avenue LaGrange Park, Illinois 60526 Phone: (800) 323-3044 (708) 352-6611 Fax: (708) 352-0499 www.ans.org Additional copies of this publication are available from: Curran Associates, Inc. 57 Morehouse Lane Red Hook, NY 12571 USA Phone: 845-758-0400 Fax: 845-758-2634 Email: [email protected] Web: www.proceedings.com Table of Contents Monday, August 25, 2014, 8:00 A.M. ISOTOPES GENERAL PLENARY—I Session Organizers: Paul T. Dickman (ANL), Rolf Zeisler (NIST), Stephen P. LaMont (LANL) Cochairs: Dale Klein (UT System), Jong-Kyung Kim (KAERI) Economics of Radioisotope Production and Sustainability 1 Ron Cameron (OECD Nuclear Energy Agency), invited Isotope Developments in the Australian Setting 2 Adri Paterson, Karina Meredith, Quan Hua (ANSTO), invited Production Capacities of ROSATOM Isotope Complex 3 Aleksey Vakulenko (JSC Isotope), invited U.S. Department of Energy Isotope Production and Research 4 Marc A. Garland (DOE), invited Monday, August 25, 2014, 10:10 A.M. ISOTOPES GENERAL PLENARY—II Cochairs: Adi Paterson (ANSTO), Mark Peters (ANL) Stable Isotope Enrichment Methods—Historical Review and Future Trends 5 Darren Brown (Trace Sciences International), invited Radioisotopes and Radiation Technology in Industry 6 Meera Venkatesh, Patrick Brisset, Sunil Sabharwal, Agnes Safrany (IAEA), invited Transforming the Playing Field; Personalized Medicine Using Theragnostic 7 Radiopharmaceuticals Suresh C.