Global Health Care Compliance News & Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rest Lawn Memorial Park Burial Book 1
REST LAWN MEMORIAL PARK BURIAL BOOK Surname FIRST NAME BIRTH-DATE DEATH or GARD SEC BLK PLOT BURIAL DATE EN Abbey Gaither William 1939 25-Mar-1963 BET 273 A 1 Abbey Geneva Hope 10-Oct-1919 18-Oct-1942 BET 273 A 2 Ackerby Seth 08/27/1878 30-Mar-1953 HOP 14 2 Adams Bertha V. 11-Jan-1909 13-Feb-2001 ARB 136 B 10 Adams Carsen 24-Dec-1918 30-Oct-1971 BET 229 B 9 Adams Eleanor 2-Nov-1937 15-Dec-2000 BET 216 B 3 Adams Elmer C. 09/18/1888 30-Aug-1981 CED 364 3 Adams Elmer Floyd 27-Jan-1917 20-Jul-1960 CED 364 6 Adams Gary Pearl 28-Apr-1941 23-Oct-1996 BET 229 B 8 Adams James Victor 12-Mar-1901 7-Mar-1974 ARB 136 B 9 Adams Lucille 4-Dec-1920 12-Jun-2013 BET 229 B 10 Adams Mabel A. 09/14/1888 28-Nov-1984 CED 364 4 Adams Marion A. 21-Apr-1928 14-Aug-2000 ARB 136 B 7 Agee Della E. 1874 15-Apr-1951 BET 249 A 2 Agee James R. 1868 15-Jul-1936 BET 249 A 1 Aho Beatrice Thompson 18-Jun-1914 28-May-1999 BET 185 B 2 Aho Toivo 9-Mar-1908 27-Feb-1983 BET 186 B 2 Akers Bertha M. 16-May-1909 9-Oct-1910 ARB 30 0 1 Akers Izorab 02/07/1863 05/08/1889 ARB 68 0 5 Akers Jabez H. 04/15/1827 30-Sep-1909 ARB 68 0 4 Akers Mabel Hoel 1879 28-Feb-1962 ARB 30 0 3 Akers Medley E. -
Senate Vote on Trump Trial Signals an Acquittal Is Likely
P2JW027000-6-A00100-17FFFF5178F ****** WEDNESDAY,JANUARY27, 2021 ~VOL. CCLXXVII NO.21 WSJ.com HHHH $4.00 DJIA 30937.04 g 22.96 0.1% NASDAQ 13626.06 g 0.1% STOXX 600 407.70 À 0.6% 10-YR. TREAS. unch , yield 1.039% OIL $52.61 g $0.16 GOLD $1,850.70 g $4.20 EURO $1.2162 YEN 103.62 In India, Farmers’ Protest Over New Law Turns Violent Microsoft What’s News SalesRise 17%Amid Business&Finance Covid-19 icrosoftposted record Mquarterly sales under- pinned by pandemic-fueled Pandemic demand forvideogaming and accelerated adoption of itscloud-computing services Demand for cloud during the health crisis. A1 services, videogaming Walgreens Bootsnamed Starbucks operating chief fuels earnings during Rosalind Brewerasits next work-from-home era CEO,making her the only Black woman leading a BY AARON TILLEY Fortune 500 company. A1 CK J&J said it expectstore- TO MicrosoftCorp. posted re- port pivotal resultsofalarge cord quarterly sales under- clinical trial of itsCovid-19 SHUTTERS pinned by pandemic-fueled de- vaccine by early next week, A/ mand forvideogaming and as the companyposted im- I/EP accelerated adoption of its AG proved quarterly sales. B1 TY cloud-computing services dur- ing the health crisis. GE booked $4.4billion Theremote-work erahas in fourth-quarter cash HARISH STREET CLASH: Indian farmers clash with police in New Delhi on Tuesday after breaking through barriers to escape po- been a boon for Microsoft. In flow,beating itsown pro- lice-approved routes for a tractor rally that coincided with a military parade celebrating India’s Republic Day. -

8Th International Conference on Isotopes and Expo
th 2 0 1 4 8th International Conference on Isotopes and Expo Preparing for Tomorrow Sponsored by the Accelerator Applications, Preliminary Program Biology & Medicine, and Isotope & Radiation Divisions of the American Nuclear Society www.8ici.org August 24-28, 2014 Hyatt Regency-Chicago Chicago, IL SPONSORS Accelerator Applications Division Biology and Medicine Division Isotopes and Radiation Division 2 2014 International Conference on Isotopes and Expo: Preliminary Program www.ans.org Table of Contents Plenary Programs and Speakers Sponsors 2 Meeting Officials 4-5 Meeting Information and Special Events 6 Plenary Programs and Speakers 7 Meeting Schedule 8-9 Monday Technical Sessions 10-13 Tuesday Technical Sessions 14-18 Wednesday Technical Sessions 19-23 Thursday Technical Sessions 23-24 2014 8TH ICI Registration Form 25 www.8ici.org 2014 International Conference on Isotopes and Expo: Preliminary Program 3 Meeting Officials Honorary Chair: General Chair: Assistant General Chair: Myung-Chul Lee Paul T. Dickman Nigel R. Stevenson President, WCI Argonne National Laboratory Clear Vascular, Inc. President, Korean Association for Radiation Application Technical Program Co-Chair: Technical Program Co-Chair: Finance Chair: Rolf Zeisler Stephen P. LaMont James T. Tanner National Institute of Standard Los Alamos National Laboratory U.S. Food and Drug Administration and Technology (retired) Publications Co-Chair: Publications Co-Chair: International Program Director: Sam Glover Bryan P. Bednarz Gulbarshyn Bozheyeva University of Cincinnati University of Wisconsin-Madison MELE Associates, Inc. Executive Advisory Board Executive Advisory Board: Executive Advisory Board: JongKyung Kim Meera Venkatesh Ron Cameron WCI, Secretary-General IAEA OECD-NEA President, KAERI Executive Advisory Board Member: President Elect, WCI International Coordinator WCI: Ilham Y. -

2006 Annual Report Board of Directors Kathryn R
Legal Aid Society of Middle Tennessee and the Cumberlands 2006 Annual Report Board of Directors Kathryn R. Edge, President N. Houston Parks, First Vice President Susan L. Kay, Second Vice President Richard K. Evans, Third Vice President Valerie Martin, Secretary John Andrew Goddard, Treasurer John Pellegrin, Past President Charles H. Warfield, Executive Committee Member at Large Clisby Barrow John T. Blankenship Richard M. Brooks Melanie T. Cagle Robert Allen Dickens Roberta Dobbins Trudy Edwards Daniel B. Eisenstein Craig Fickling Barbara Gooch Fannie J. Harris Amy T. Hollars G. Wilson Horde Lou Lavender Turner McCullough, Jr. James D. Petersen Teresa Poston Adrie Mae Rhodes Steve Rhodey Denice Scott Keith S. Smartt Gregory D. Smith Guilford F. Thorton, Jr. James L. Weatherly, Jr. Shelby York Nashville Pro Bono Program Board Mary Griffin, Chair Andrée Blumstein Daniel B. Eisenstein Richard Green Tonya Mitchem Grindon Susan L. Kay N. Sue Van Sant Palmer Jonathan E. Richardson Robyn L. Ryan Thor Y. Urness Mark H. Westlake Message From the President of the Board Dear Friends and Colleagues: Legal Aid Society provides a helping hand to those who need it most – low-income citizens who cannot afford professional legal help and have nowhere to turn. When they receive the help they need, when they learn that there is justice out there for everyone, then the entire community benefits. America was founded on the ideal that everyone should be treated equally under the law. And while it is still possible to find examples of justice best serving those who can afford to pay for it, Legal Aid proves that the ideals which are fundamental to a fair and just society do, indeed, exist in our nation today. -

The CEO Action for Diversity & Inclusion™ Aims to Rally the Business Community to Advance Diversity & Inclusion Within
The CEO Action for Diversity & Inclusion™ aims to rally the business community to advance diversity & inclusion within the workplace by working collectively across organizations and sectors. It outlines a specific set of actions the undersigned companies will take to cultivate a trusting environment where all ideas are welcomed and employees feel comfortable and empowered to discuss diversity & inclusion. All the signatories serve as leaders of their companies and have committed to implementing the following pledge within their workplaces. Where companies have already implemented one or several of the commitments, the undersigned commit to support other companies in doing the same. The persistent inequities across our country underscore our urgent, national need to address and alleviate racial, ethnic and other tensions and to promote diversity within our communities. As leaders of some of America’s largest corporations, we manage thousands of employees and play a critical role in ensuring that inclusion is core to our workplace culture and that our businesses are representative of the communities we serve. Moreover, we know that diversity is good for the economy; it improves corporate performance, drives growth and enhances employee engagement. Simply put, organizations with diverse teams perform better. We recognize that diversity & inclusion are multifaceted issues and that we need to tackle these subjects holistically to better engage and support all underrepresented groups within business. To do this, we believe we also need to address honestly and head-on the concerns and needs of our diverse employees and increase equity for all, including Blacks, Latinos, Asians, Native Americans, LGBTQ, disabled, veterans and women. -

1980 Surname
Surname Given Age Date Page Maiden Note Abbett Howard 91 6-Jan D-2 Abercrombie Levi Sr. 30-Mar E-15 Able Cora Ree 73 4-Dec C-8 Acevedo Marcelina P. 68 11-Dec D-2 Acton John Wesley 85 12-May D-1 Adam Michael Sr. 88 7-Mar B-3 Adam Millee 59 3-Mar B-6 Adam Sophie (Sister Ann 66 15-Dec B-7 Madeline) Adamczyk Josephine 82 4-May D-6 Adams Francis (Sheik) 78 14-Jul C-5 Adams Ruth Carol 41 17-Oct C-5 Adams William H. 63 21-Aug B-2 Ade Eleanor Anne 63 17-Jul C-14 Adelsperger James F. 78 22-Dec C-5 Adkins Otis C. 67 24-Jun C-1 Adler Florian F. 83 10-Aug D-2 Afflek Day Malo 18-Mar B-4 Agosto Gregorio 56 3-Dec E-2 Ahlborn Rudolph C. 81 10-Sep E-1 Ahlborn Walter W. 78 20-Jul D-1 Aikman Myrtle M. 74 26-Nov D-1 Albert Joseph (Larry) 51 11-Jan B-4 Albertson Russell A. 83 27-Jul C-7 Alderden Gertrude 76 7-Jul B-4 Aleman Sadie 21-Jan B-5 Alexander Edward L. 46 5-Aug C-3 Alexander Robert W. Jr. 65 2-Jan C-9 Alexander Sonja E. 67 11-Feb C-4 Alier Audra 65 30-Nov E-11 Brown Allen Clarence F. 57 9-May B-5 Allen Norman 79 7-Aug C-4 Picture included. Allen Rabe (Ray) 95 22-Dec C-5 Allen William J. -
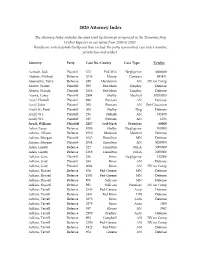
2020 Attorney Index
2020 Attorney Index The Attorney Index includes the cases tried by attorneys as reported in the Tennessee Jury Verdict Reporter in our issues from 2005 to 2020. Results are sorted alphabetically and then include the party represented, case index number, jurisdiction and verdict. Attorney Party Case No.-County Case Type Verdict Aamodt, Jodi Plaintiff 372 Fed-Win Negligence 8000000 Abelow, Michael Defense 2538 Maury Contract 995431 Abernathy, Terry Defense 240 Hardeman AN DV on Comp Abioto, Walter Plaintiff 508 Fed-Mem Employ Defense Abioto, Wanda Plaintiff 2618 Fed-Mem Employ Defense Acerra, Carey Plaintiff 2894 Shelby Medical 30035000 Acuff, Howell Plaintiff 920 Putnam AN Defense Acuff, John Plaintiff 292 Putnam AN Def-Causation Acuff, Jr., Fred Plaintiff 500 Shelby Dog Defense Acuff, W.I. Plaintiff 296 DeKalb AN 185100 Acuff, W.I. Plaintiff 285 Putnam AN 1233 Acuff, William Plaintiff 3257 Fed-Nash Premises 68000 Adair, Lacey Defense 1095 Shelby Negligence 100000 Adams, Allison Defense 2510 Madison Medical Defense Adams, Morgan Plaintiff 1625 Hamilton MN Defense Adams, Morgan Plaintiff 2068 Hamilton AN 9250000 Aden, Gareth Defense 522 Hamilton FELA 5000000 Aden, Gareth Defense 1368 Hamilton FELA 3000000 Adkins, Gary Plaintiff 216 Knox Negligence 132000 Adkins, Gary Plaintiff 838 Knox AN Defense Adkins, Gary Plaintiff 2084 Knox AN DV on Comp Adkins, Russell Defense 678 Fed-Greene MN Defense Adkins, Russell Defense 1503 Fed-Greene MN Defense Adkins, Russell Defense 976 Sullivan MN Defense Adkins, Russell Defense 981 Sullivan Premises DV -

Oumaimamajiti 11047488 Scrip
Are the Sleeping Giants Awakening? An Investigation into the Investment Stewardship Efforts of the Big Three Passive Investment Managers O. Majiti Student number: 11047488 27th June 2018 Bachelor Thesis in Political Science University of Amsterdam Supervisor: Dr. E.M. Heemskerk Second reader: Dr. J.R. Fichtner Acknowledgements The thesis that lies in front of you is the conclusion of both my bachelor’s in political science and time at the CORPNET research group. Firstly, I would like to express my thanks to my supervisor, Eelke Heemskerk, for guiding me through the process of writing this thesis and giving me the opportunity to get a taste of social science research even before graduating by inviting me to join the CORPNET group. Also, I would also like to thank the CORPNET team members, Frank, Jan, Diliara, Javier, Milan, Jouke and Dawid, for the (research) skills they have taught me; the advice and feedback they were always happy to give and mostly for enabling an amiable and fruitful working environment. Further, I would like to express special thanks to my parents for always believing in me and providing the love and support that have enabled me to achieve my potential. Also, I would like to thank my sister, Btissame, for inspiring me to fight hard for what I want, no matter what others think, and for always being there to put things into perspective. My final words of gratitude are directed towards my other sisters, Chaymae, Nada and Hiba, for their endless encouragements, for always sympathizing with me and for never letting life get boring or dull. -

WE STAND for DEMOCRACY. a Government of the People, by the People
A14 EZ RE the washington post . wednesday, april 14, 2021 EZ RE A15 ADVERTISEMENT ADVERTISEMENT WE STAND FOR DEMOCRACY. A Government of the people, by the people. A beautifully American ideal, but a reality denied to many for much of this nation’s history. As Americans, we know that in our democracy we should not expect to agree on everything. However, regardless of our political affiliations, we believe the very foundation of our electoral process rests upon the ability of each of us to cast our ballots for the candidates of our choice. For American democracy to work for any of us, we must ensure the right to vote for all of us. We all should feel a responsibility to defend the right to vote and to oppose any discriminatory legislation or measures that restrict or prevent any eligible voter from having an equal and fair opportunity to cast a ballot. Voting is the lifeblood of our democracy and we call upon all Americans to join us in taking a nonpartisan stand for this most basic and fundamental right of all Americans. Paid for by: Ursula Burns, Debra Lee, Ken Jacobs, Joel Cutler, David Fialkow, Hemant Taneja, Casey Wasserman, Ken Chenault, Ken Frazier, William Lewis, Clarence Otis, Charles Phillips blackeconomicalliance.org Email: [email protected] Original Signatories Cambridge Associates Individuals Roger Crandall, Chairman, President The founders of Tango Fritz Lanman Kieran O’Reilly & Rory O’Reilly, Daniel Schreiber & Shai Wininger, John Zimmer, Peter Fader, Professor of Marketing,The & Chief Executive Officer, MassMutual Co-founders, Millions cofounders, Lemonade Co-founder & President, Lyft Rodney C. -

Larry Fink CEO Letter | Blackrock Blackrock Ishares Aladdin Our Company About Us Newsroom Insights Investor Relations Corporate Sustainability Careers Local Websites
Larry Fink CEO Letter | BlackRock BlackRock iShares Aladdin Our company About Us Newsroom Insights Investor Relations Corporate sustainability Careers Local websites Read Larry Fink’s 2021 letter to CEOs Larry Fink's 2021 letter to CEOs Dear CEO, BlackRock is a fiduciary to our clients, helping them invest for long-term goals. Most of the money we manage is for retirement – for individuals and pension beneficiaries like teachers, firefighters, doctors, businesspeople, and many others. It is their money we manage, not our own. The trust our clients place in us, and our role as the link between our clients and the companies they invest in, gives us a great responsibility to advocate on their behalf. This is why I write to you each year, seeking to highlight issues that are pivotal to creating durable value – issues such as capital management, long-term strategy, purpose, and climate change. We have long believed that our clients, as shareholders Read BlackRock'sin your company, 2021 willletter benefit to clients if you can create enduring, sustainable value for all of your stakeholders. I began writing these letters in the wake of the financial crisis. But over the past year, we experienced something even more far-reaching – a pandemic that has enveloped the entire globe and changed it permanently. It has both exacted a horrific human toll and transformed the way we live – the way we work, learn, access medicine, and much more. https://www.blackrock.com/corporate/investor-relations/larry-fink-ceo-letter[1/27/2021 1:14:11 PM] Larry Fink CEO Letter | BlackRock The consequences of the pandemic have been highly uneven. -

2019 Investment Stewardship Annual Report
2019 Investment Stewardship Annual Report August 2019 Annual Report Navigating long-term change – 3 Active the year in review 2018-2019 Investment Stewardship 4 stewardship: highlights creating long- Our achievements 5 Our principles, guidelines, priorities, 7 term value and commentaries The Investment Stewardship Engagement and voting case studies 10-22 Annual Report provides an • Board quality and effectiveness remain overview of BlackRock’s approach our primary focus • Corporate strategy and capital allocation to corporate governance and • Executive compensation stewardship in support of long- • Environmental risk and opportunities term value creation for our clients. • Human capital management as an In this report we provide practical investment issue examples of the BlackRock Spotlight on activism 23 Investment Stewardship (BIS) Engagement and voting statistics 24 team’s work over the year, Investor perspective and public policy 25 distilling some of the trends and Industry affiliations and memberships 28 company-specific situations reported in our regional quarterly Appendix reports. We emphasize the List of companies engaged 31 outcome of our engagements with BlackRock’s 2019 PRI assessment 38 companies, including some which report and score have spanned several years. We also provide examples of where we have contributed to the public discourse on corporate Our Annual Report reporting period is July 1, 2018 to June 30, 2019, representing the Securities and Exchange governance and investment Commission’s (SEC) 12-month reporting period for US mutual funds, including iShares. stewardship. Navigating long-term change – the year in review The adage “change is the only constant” has never been more true than in the past year. -

Wall Street's Border Wall
The Partnership for Working Families is a national network of 17 powerful city and regional affiliate groups based in major urban areas across the country. The Partnership advocates for and supports policies and movements that help build more just and sustainable communities where we live and work. Taking lessons learned at the local level, the Partnership applies them to the national conversation to build a framework for addressing climate change, inequality, racial and social injustice. The Center for Popular Democracy (CPD) works to create equity, opportunity and a dynamic democracy in partnership with high-impact base-building organizations, organizing alliances, and progressive unions. CPD strengthens our collective capacity to envision and win an innovative pro-worker, pro- immigrant, racial and economic justice agenda. New York Communities for Change (NYCC) is a multi-racial membership based organization of working families fighting against economic and racial oppression. NYCC members are agents of change, building movements and campaigns from the ground up and fighting corporate power at its core. NYCC members use direct action to defend & uplift our communities, challenge capital, and fight back against racist structures and economic policies that continue to extract wealth from our communities and neighborhoods. Make the Road New York (MRNY) builds the power of Latino and working class communities to achieve dignity and justice through organizing, policy innovation, transformative education, and survival services. MRNY is the largest grassroots community organization in New York offering services and organizing the immigrant community, with more than 20,000 members and community centers in Brooklyn, Queens, Staten Island, and Long Island.