Pointing the Finger
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Table 2. Functional Classification of Genes Differentially Regulated After HOXB4 Inactivation in HSC/Hpcs
Table 2. Functional classification of genes differentially regulated after HOXB4 inactivation in HSC/HPCs Symbol Gene description Fold-change (mean ± SD) Signal transduction Adam8 A disintegrin and metalloprotease domain 8 1.91 ± 0.51 Arl4 ADP-ribosylation factor-like 4 - 1.80 ± 0.40 Dusp6 Dual specificity phosphatase 6 (Mkp3) - 2.30 ± 0.46 Ksr1 Kinase suppressor of ras 1 1.92 ± 0.42 Lyst Lysosomal trafficking regulator 1.89 ± 0.34 Mapk1ip1 Mitogen activated protein kinase 1 interacting protein 1 1.84 ± 0.22 Narf* Nuclear prelamin A recognition factor 2.12 ± 0.04 Plekha2 Pleckstrin homology domain-containing. family A. (phosphoinosite 2.15 ± 0.22 binding specific) member 2 Ptp4a2 Protein tyrosine phosphatase 4a2 - 2.04 ± 0.94 Rasa2* RAS p21 activator protein 2 - 2.80 ± 0.13 Rassf4 RAS association (RalGDS/AF-6) domain family 4 3.44 ± 2.56 Rgs18 Regulator of G-protein signaling - 1.93 ± 0.57 Rrad Ras-related associated with diabetes 1.81 ± 0.73 Sh3kbp1 SH3 domain kinase bindings protein 1 - 2.19 ± 0.53 Senp2 SUMO/sentrin specific protease 2 - 1.97 ± 0.49 Socs2 Suppressor of cytokine signaling 2 - 2.82 ± 0.85 Socs5 Suppressor of cytokine signaling 5 2.13 ± 0.08 Socs6 Suppressor of cytokine signaling 6 - 2.18 ± 0.38 Spry1 Sprouty 1 - 2.69 ± 0.19 Sos1 Son of sevenless homolog 1 (Drosophila) 2.16 ± 0.71 Ywhag 3-monooxygenase/tryptophan 5- monooxygenase activation protein. - 2.37 ± 1.42 gamma polypeptide Zfyve21 Zinc finger. FYVE domain containing 21 1.93 ± 0.57 Ligands and receptors Bambi BMP and activin membrane-bound inhibitor - 2.94 ± 0.62 -

Functional Roles of Bromodomain Proteins in Cancer
cancers Review Functional Roles of Bromodomain Proteins in Cancer Samuel P. Boyson 1,2, Cong Gao 3, Kathleen Quinn 2,3, Joseph Boyd 3, Hana Paculova 3 , Seth Frietze 3,4,* and Karen C. Glass 1,2,4,* 1 Department of Pharmaceutical Sciences, Albany College of Pharmacy and Health Sciences, Colchester, VT 05446, USA; [email protected] 2 Department of Pharmacology, Larner College of Medicine, University of Vermont, Burlington, VT 05405, USA; [email protected] 3 Department of Biomedical and Health Sciences, University of Vermont, Burlington, VT 05405, USA; [email protected] (C.G.); [email protected] (J.B.); [email protected] (H.P.) 4 University of Vermont Cancer Center, Burlington, VT 05405, USA * Correspondence: [email protected] (S.F.); [email protected] (K.C.G.) Simple Summary: This review provides an in depth analysis of the role of bromodomain-containing proteins in cancer development. As readers of acetylated lysine on nucleosomal histones, bromod- omain proteins are poised to activate gene expression, and often promote cancer progression. We examined changes in gene expression patterns that are observed in bromodomain-containing proteins and associated with specific cancer types. We also mapped the protein–protein interaction network for the human bromodomain-containing proteins, discuss the cellular roles of these epigenetic regu- lators as part of nine different functional groups, and identify bromodomain-specific mechanisms in cancer development. Lastly, we summarize emerging strategies to target bromodomain proteins in cancer therapy, including those that may be essential for overcoming resistance. Overall, this review provides a timely discussion of the different mechanisms of bromodomain-containing pro- Citation: Boyson, S.P.; Gao, C.; teins in cancer, and an updated assessment of their utility as a therapeutic target for a variety of Quinn, K.; Boyd, J.; Paculova, H.; cancer subtypes. -

Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin
cells Review Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin Agnieszka Bochy ´nska,Juliane Lüscher-Firzlaff and Bernhard Lüscher * ID Institute of Biochemistry and Molecular Biology, Medical School, RWTH Aachen University, Pauwelsstrasse 30, 52057 Aachen, Germany; [email protected] (A.B.); jluescher-fi[email protected] (J.L.-F.) * Correspondence: [email protected]; Tel.: +49-241-8088850; Fax: +49-241-8082427 Received: 18 January 2018; Accepted: 27 February 2018; Published: 2 March 2018 Abstract: Regulation of gene expression is achieved by sequence-specific transcriptional regulators, which convey the information that is contained in the sequence of DNA into RNA polymerase activity. This is achieved by the recruitment of transcriptional co-factors. One of the consequences of co-factor recruitment is the control of specific properties of nucleosomes, the basic units of chromatin, and their protein components, the core histones. The main principles are to regulate the position and the characteristics of nucleosomes. The latter includes modulating the composition of core histones and their variants that are integrated into nucleosomes, and the post-translational modification of these histones referred to as histone marks. One of these marks is the methylation of lysine 4 of the core histone H3 (H3K4). While mono-methylation of H3K4 (H3K4me1) is located preferentially at active enhancers, tri-methylation (H3K4me3) is a mark found at open and potentially active promoters. Thus, H3K4 methylation is typically associated with gene transcription. The class 2 lysine methyltransferases (KMTs) are the main enzymes that methylate H3K4. KMT2 enzymes function in complexes that contain a necessary core complex composed of WDR5, RBBP5, ASH2L, and DPY30, the so-called WRAD complex. -

Histone-Binding of DPF2 Mediates Its Repressive Role in Myeloid Differentiation
Histone-binding of DPF2 mediates its repressive role in myeloid differentiation Ferdinand M. Hubera,1, Sarah M. Greenblattb,1, Andrew M. Davenporta,1, Concepcion Martinezb,YeXub,LyP.Vuc, Stephen D. Nimerb,2, and André Hoelza,2 aDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125; bSylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL 33136; and cMolecular Pharmacology Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065 Edited by Douglas C. Rees, Howard Hughes Medical Institute, California Institute of Technology, Pasadena, CA, and approved April 26, 2017 (received for review January 6, 2017) Double plant homeodomain finger 2 (DPF2) is a highly evolution- RUNX1 form a methylation-dependent repressive complex in arily conserved member of the d4 protein family that is ubiqui- AML, although it remains unclear whether the two proteins bind tously expressed in human tissues and was recently shown to each other directly or act concertedly as part of a larger complex. inhibit the myeloid differentiation of hematopoietic stem/progen- Here, we present the crystal structure of the human DPF2 itor and acute myelogenous leukemia cells. Here, we present the tandem PHD finger domain at a 1.6-Å resolution. We demon- crystal structure of the tandem plant homeodomain finger domain strate that the DPF2 tandem PHD finger domain binds acetylated of human DPF2 at 1.6-Å resolution. We show that DPF2 interacts H3 and H4 histone tails, identify the primary determinants of with the acetylated tails of both histones 3 and 4 via bipartite histone recognition, and confirm these interactions in vivo. -

Genomic Analyses Reveal Foxg As an Upstream Regulator of Wnt1 Required For
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.08.416008; this version posted December 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genomic analyses reveal FoxG as an upstream regulator of wnt1 required for posterior identity specification in planarians E. Pascual-Carreras1, M. Marín-Barba3, S. Castillo-Lara1, P. Coronel-Córdoba1, M.S. Magri2, G.N. Wheeler3 J.F. Abril1, J.L. Gomez-Skarmeta2, E. Saló1* & T. Adell1* 1 Department of Genetics, Microbiology and Statistics, Universitat de Barcelona (UB) & Institute of Biomedicine of Universitat de Barcelona (IBUB), Barcelona, Spain. 2 Centro Andaluz de Biología del Desarollo (CABD), Universidad Pablo de Olavide, Sevilla, Spain. 3 School of Biological Sciences, University of East Anglia, Norwich Research Park, Norwich, UK. *Corresponding authors: Emili Saló ([email protected]) and Teresa Adell ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.12.08.416008; this version posted December 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Embryonic specification of the first body axis requires the formation of an Organizer, a group of cells with the ability to instruct fates in the surrounding tissue. The existence of organizing regions in adults, i.e. -

Epigenetic Mechanisms of Lncrnas Binding to Protein in Carcinogenesis
cancers Review Epigenetic Mechanisms of LncRNAs Binding to Protein in Carcinogenesis Tae-Jin Shin, Kang-Hoon Lee and Je-Yoel Cho * Department of Biochemistry, BK21 Plus and Research Institute for Veterinary Science, School of Veterinary Medicine, Seoul National University, Seoul 08826, Korea; [email protected] (T.-J.S.); [email protected] (K.-H.L.) * Correspondence: [email protected]; Tel.: +82-02-800-1268 Received: 21 September 2020; Accepted: 9 October 2020; Published: 11 October 2020 Simple Summary: The functional analysis of lncRNA, which has recently been investigated in various fields of biological research, is critical to understanding the delicate control of cells and the occurrence of diseases. The interaction between proteins and lncRNA, which has been found to be a major mechanism, has been reported to play an important role in cancer development and progress. This review thus organized the lncRNAs and related proteins involved in the cancer process, from carcinogenesis to metastasis and resistance to chemotherapy, to better understand cancer and to further develop new treatments for it. This will provide a new perspective on clinical cancer diagnosis, prognosis, and treatment. Abstract: Epigenetic dysregulation is an important feature for cancer initiation and progression. Long non-coding RNAs (lncRNAs) are transcripts that stably present as RNA forms with no translated protein and have lengths larger than 200 nucleotides. LncRNA can epigenetically regulate either oncogenes or tumor suppressor genes. Nowadays, the combined research of lncRNA plus protein analysis is gaining more attention. LncRNA controls gene expression directly by binding to transcription factors of target genes and indirectly by complexing with other proteins to bind to target proteins and cause protein degradation, reduced protein stability, or interference with the binding of other proteins. -
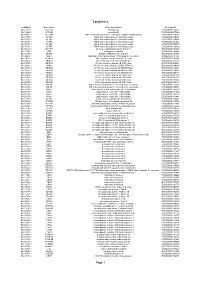
Epigenetics Page 1
Epigenetics esiRNA ID Gene Name Gene Description Ensembl ID HU-13237-1 ACTL6A actin-like 6A ENSG00000136518 HU-13925-1 ACTL6B actin-like 6B ENSG00000077080 HU-14457-1 ACTR1A ARP1 actin-related protein 1 homolog A, centractin alpha (yeast) ENSG00000138107 HU-10579-1 ACTR2 ARP2 actin-related protein 2 homolog (yeast) ENSG00000138071 HU-10837-1 ACTR3 ARP3 actin-related protein 3 homolog (yeast) ENSG00000115091 HU-09776-1 ACTR5 ARP5 actin-related protein 5 homolog (yeast) ENSG00000101442 HU-00773-1 ACTR6 ARP6 actin-related protein 6 homolog (yeast) ENSG00000075089 HU-07176-1 ACTR8 ARP8 actin-related protein 8 homolog (yeast) ENSG00000113812 HU-09411-1 AHCTF1 AT hook containing transcription factor 1 ENSG00000153207 HU-15150-1 AIRE autoimmune regulator ENSG00000160224 HU-12332-1 AKAP1 A kinase (PRKA) anchor protein 1 ENSG00000121057 HU-04065-1 ALG13 asparagine-linked glycosylation 13 homolog (S. cerevisiae) ENSG00000101901 HU-13552-1 ALKBH1 alkB, alkylation repair homolog 1 (E. coli) ENSG00000100601 HU-06662-1 ARID1A AT rich interactive domain 1A (SWI-like) ENSG00000117713 HU-12790-1 ARID1B AT rich interactive domain 1B (SWI1-like) ENSG00000049618 HU-09415-1 ARID2 AT rich interactive domain 2 (ARID, RFX-like) ENSG00000189079 HU-03890-1 ARID3A AT rich interactive domain 3A (BRIGHT-like) ENSG00000116017 HU-14677-1 ARID3B AT rich interactive domain 3B (BRIGHT-like) ENSG00000179361 HU-14203-1 ARID3C AT rich interactive domain 3C (BRIGHT-like) ENSG00000205143 HU-09104-1 ARID4A AT rich interactive domain 4A (RBP1-like) ENSG00000032219 HU-12512-1 ARID4B AT rich interactive domain 4B (RBP1-like) ENSG00000054267 HU-12520-1 ARID5A AT rich interactive domain 5A (MRF1-like) ENSG00000196843 HU-06595-1 ARID5B AT rich interactive domain 5B (MRF1-like) ENSG00000150347 HU-00556-1 ASF1A ASF1 anti-silencing function 1 homolog A (S. -

Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis
International Journal of Molecular Sciences Review Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis Elzbieta Poreba 1,* , Krzysztof Lesniewicz 2 and Julia Durzynska 1,* 1 Institute of Experimental Biology, Faculty of Biology, Adam Mickiewicz University, ul. Uniwersytetu Pozna´nskiego6, 61-614 Pozna´n,Poland 2 Department of Molecular and Cellular Biology, Institute of Molecular Biology and Biotechnology, Faculty of Biology, Adam Mickiewicz University, ul. Uniwersytetu Pozna´nskiego6, 61-614 Pozna´n,Poland; [email protected] * Correspondence: [email protected] (E.P.); [email protected] (J.D.); Tel.: +48-61-829-5857 (E.P.) Received: 19 November 2020; Accepted: 6 December 2020; Published: 8 December 2020 Abstract: KMT2 (histone-lysine N-methyltransferase subclass 2) complexes methylate lysine 4 on the histone H3 tail at gene promoters and gene enhancers and, thus, control the process of gene transcription. These complexes not only play an essential role in normal development but have also been described as involved in the aberrant growth of tissues. KMT2 mutations resulting from the rearrangements of the KMT2A (MLL1) gene at 11q23 are associated with pediatric mixed-lineage leukemias, and recent studies demonstrate that KMT2 genes are frequently mutated in many types of human cancers. Moreover, other components of the KMT2 complexes have been reported to contribute to oncogenesis. This review summarizes the recent advances in our knowledge of the role of KMT2 complexes in cell transformation. In addition, it discusses the therapeutic targeting of different components of the KMT2 complexes. Keywords: histone–lysine N-methyltransferase 2; COMPASS; COMPASS-like; H3K4 methylation; oncogenesis; cancer; epigenetics; chromatin 1. -

Bptf Is Essential for Murine Neocortical Development
Bptf is essential for murine neocortical development Gerardo Zapata Thesis submitted to the Faculty of Graduate and Postdoctoral Studies in partial fulfillment of the requirements for the Master’s degree in Biochemistry with specialization in Bioinformatics Department of Biochemistry, Microbiology and Immunology Faculty of Medicine University of Ottawa © Gerardo Zapata, Ottawa, Canada, 2020 Abstract Chromatin remodeling complexes modulate DNA accessibility permitting neuronal progenitor cells to proliferate and differentiate to form the mammalian neocortex. In the case of BPTF (Bromodomain PHD transcription Factor), the major subunit of a chromatin remodelling complex called NURF (Nucleosome Remodelling Factor), mutations leading to its haploinsufficiency have been linked to cause a recently annotated human neurodevelopmental disorder called NEDDFL (Neurodevelopmental disorder with dysmorphic facies and distal limb anomalies). Patients with this syndrome are mainly characterized with microcephaly and intellectual disability. We conditionally knockout (cKO) the Bptf gene during neocortical neurogenesis to analyze its role during embryonic and postnatal brain development. The Bptf cKO animals reveal significant forebrain hypoplasia. During cortical neurogenesis, the Bptf cKO mice show a reduction in intermediate neuronal progenitor (INP) cells, an increase in apoptosis as well as a prolonged cell cycle within proliferating progenitors. Similarly, the postmitotic pyramidal neurons of the Bptf cKO mice contained lower levels of Ctip2 and Foxp1. Lastly, our RNA-seq analysis delineated gene pathways deregulated by Bptf removal, which are involved in neurogenesis and neuronal differentiation. Our results indicate that Bptf is critical for murine telencephalon neurogenesis. The hypoplasia demonstrated in the mouse model can resemble the microcephaly displayed by the human NEDDFL patients, highlighting the relevance of chromatin remodelling complexes during intricate neural developmental processes. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

The Prevalence of Nucleic Acid Binding Among Reader Domains
molecules Review Reading More than Histones: The Prevalence of Nucleic Acid Binding among Reader Domains Tyler M. Weaver †, Emma A. Morrison † and Catherine A. Musselman * Department of Biochemistry, Carver College of Medicine, University of Iowa, Iowa City, IA 52242, USA; [email protected] (T.M.W.); [email protected] (E.A.M.) * Correspondence: [email protected] † These authors contributed equally to this work. Received: 1 September 2018; Accepted: 7 October 2018; Published: 12 October 2018 Abstract: The eukaryotic genome is packaged into the cell nucleus in the form of chromatin, a complex of genomic DNA and histone proteins. Chromatin structure regulation is critical for all DNA templated processes and involves, among many things, extensive post-translational modification of the histone proteins. These modifications can be “read out” by histone binding subdomains known as histone reader domains. A large number of reader domains have been identified and found to selectively recognize an array of histone post-translational modifications in order to target, retain, or regulate chromatin-modifying and remodeling complexes at their substrates. Interestingly, an increasing number of these histone reader domains are being identified as also harboring nucleic acid binding activity. In this review, we present a summary of the histone reader domains currently known to bind nucleic acids, with a focus on the molecular mechanisms of binding and the interplay between DNA and histone recognition. Additionally, we highlight the functional implications of nucleic acid binding in chromatin association and regulation. We propose that nucleic acid binding is as functionally important as histone binding, and that a significant portion of the as yet untested reader domains will emerge to have nucleic acid binding capabilities. -

Bioinformatics Analysis for the Identification of Differentially Expressed Genes and Related Signaling Pathways in H
Bioinformatics analysis for the identification of differentially expressed genes and related signaling pathways in H. pylori-CagA transfected gastric cancer cells Dingyu Chen*, Chao Li, Yan Zhao, Jianjiang Zhou, Qinrong Wang and Yuan Xie* Key Laboratory of Endemic and Ethnic Diseases , Ministry of Education, Guizhou Medical University, Guiyang, China * These authors contributed equally to this work. ABSTRACT Aim. Helicobacter pylori cytotoxin-associated protein A (CagA) is an important vir- ulence factor known to induce gastric cancer development. However, the cause and the underlying molecular events of CagA induction remain unclear. Here, we applied integrated bioinformatics to identify the key genes involved in the process of CagA- induced gastric epithelial cell inflammation and can ceration to comprehend the potential molecular mechanisms involved. Materials and Methods. AGS cells were transected with pcDNA3.1 and pcDNA3.1::CagA for 24 h. The transfected cells were subjected to transcriptome sequencing to obtain the expressed genes. Differentially expressed genes (DEG) with adjusted P value < 0.05, | logFC |> 2 were screened, and the R package was applied for gene ontology (GO) enrichment and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The differential gene protein–protein interaction (PPI) network was constructed using the STRING Cytoscape application, which conducted visual analysis to create the key function networks and identify the key genes. Next, the Submitted 20 August 2020 Kaplan–Meier plotter survival analysis tool was employed to analyze the survival of the Accepted 11 March 2021 key genes derived from the PPI network. Further analysis of the key gene expressions Published 15 April 2021 in gastric cancer and normal tissues were performed based on The Cancer Genome Corresponding author Atlas (TCGA) database and RT-qPCR verification.