Pentosan Polysulfate Sodium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Intravesical Cocktails PBS/IC
International Painful Bladder Foundation Interstitial Cystitis/Painful Bladder Syndrome Anaesthetic intravesical cocktails 1. Anaesthetic cocktail – Robert Moldwin, MD 1:1 mixture of 0.5% Marcaine and 2% Lidocaine jelly – about 40 cc total. To this solution are added: Heparin sulphate 10,000 IU Triamcinolone 40 mg Gentamycin 80 mg or a post-procedural prophylactic antibiotic. Administration: Patients are instructed to hold the solution for about 30 minutes, then to void. When given as a diagnostic test, patients will generally sense relief of pain within 5-10 minutes. The only (rare) problems that we’ve encountered are the following: Patients may experience “rebound” pain once the solution has worn off (within 3-5 hours). This generally resolves with continued instillations. When given as therapy, we usually administer the cocktail on a weekly basis for 8-12 weeks. This is the length of time usually needed to get a prolonged response. Then, the duration between instillations is increased to q 2 weeks to q 3 weeks, etc., ultimately with the goal of discontinuance. Patients may experience urinary retention requiring catheterization. This seems to be particularly a problem in patients who appear to have pre-existing voiding dysfunction, those patients who initially present with a poor urinary flow rate, an interrupted urinary stream, etc. The urinary retention can usually be circumvented by delivering a lower total volume. 2. Marcaine with steroid cocktail – Nagendra Mishra, MD Marcaine 40 ml Heparin sulphate 10,000 IU Dexamethasone 2 cc Sodium bicarbonate 20 ml Administration: This cocktail should be held in the bladder for 20 minutes. It should be administered every 15 days for a total of 6 treatments and then as needed. -

Evaluation of Pentosan Polysulfate Sodium in the Postoperative Recovery from Cranial Cruciate Injury in Dogs: a Randomized
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/6367210 Evaluation of Pentosan Polysulfate Sodium in the Postoperative Recovery from Cranial Cruciate Injury in Dogs: A Randomized... Article in Veterinary Surgery · May 2007 DOI: 10.1111/j.1532-950X.2007.00256.x · Source: PubMed CITATIONS READS 16 103 4 authors, including: Steven C Budsberg Mary Sarah Bergh University of Georgia Iowa State University 129 PUBLICATIONS 3,106 CITATIONS 22 PUBLICATIONS 366 CITATIONS SEE PROFILE SEE PROFILE All content following this page was uploaded by Steven C Budsberg on 11 March 2014. The user has requested enhancement of the downloaded file. Veterinary Surgery 36:234–244, 2007 Evaluation of Pentosan Polysulfate Sodium in the Postoperative Recovery from Cranial Cruciate Injury in Dogs: A Randomized, Placebo-Controlled Clinical Trial STEVEN C. BUDSBERG, DVM, MS, Diplomate ACVS, MARY SARAH BERGH, DVM, LISA R. REYNOLDS, BS, and HEATHER K. STREPPA, DVM, Diplomate ACVS Objective—To evaluate the efficacy of pentosan polysulfate (PPS) for improving the recovery period and mitigate the progression of osteoarthritis (OA) of the canine stifle after extracapsular stabilization of cranial cruciate ligament (CCL) injuries. Study Design—Randomized, blinded, placebo-controlled clinical trial. Animals—Dogs (n ¼ 40) with unilateral CCL instability. Methods—Each dog had an extracapsular stabilization of the stifle with or without partial men- iscectomy. Dogs were divided into 4 groups based on preoperative radiographic assessment and whether a partial meniscectomy was performed. Dogs were randomly assigned to either (3 mg/kg) PPS or placebo treatment in each group, and then injected subcutaneously weekly for 4 weeks. -
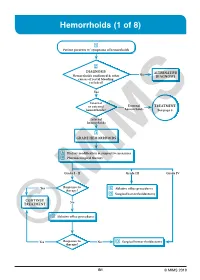
Hemorrhoids (1 of 8)
Hemorrhoids (1 of 8) 1 Patient presents w/ symptoms of hemorrhoids 2 DIAGNOSIS No ALTERNATIVE Hemorrhoids confi rmed & other DIAGNOSIS causes of rectal bleeding excluded? Yes Internal or external External TREATMENT hemorrhoids? hemorrhoids See page 3 Internal hemorrhoids 3 GRADE HEMORRHOIDS A Dietary modifi cation & supportive measures B Pharmacological therapy Grade I - II Grade III Grade IV Yes Response to C Ablative offi ce procedures therapy? D Surgical hemorrhoidectomy CONTINUE No TREATMENT C Ablative offi ce procedures Yes Response to No D Surgical hemorrhoidectomy ©therapy? MIMS B1 © MIMS 2019 Hemorrhoids (2 of 8) 1 SYMPTOMS ATTRIBUTED TO HEMORRHOIDS • Rectal bleeding - Most common presenting symptom - Bright red blood which may drip or squirt into the toilet bowl or scanty amounts may be seen on toilet tissue • Discomfort due to rectal protrusion or lump • Anal pain • HEMORRHOIDS Anal itching 2 DIAGNOSIS Medical History • Assess nature, duration & severity of symptoms - Ask about bleeding, its amount & frequency - Ask about presence of prolapsing tissue, its timing & reproducibility • Elicit possible risk factors for development of hemorrhoidal symptoms - Low-fi ber diets cause small-caliber stools, resulting in straining during defecation & engorgement of hemorrhoids - Prolonged sitting on a toilet which may cause a problem in the venous return in the perianal area - Pregnancy - Advanced age • Th e signs & symptoms of hemorrhoids are not specifi c to the disease, so care must be taken to avoid missing other causes of pathology • Obtain -

Management of Joint Disease in the Sport Horse
1 MANAGEMENT OF JOINT DISEASE IN THE SPORT HORSE Management of Joint Disease in the Sport Horse C. WAYNE MCILWRAITH Colorado State University, Ft. Collins, Colorado INTRODUCTION The joint is an organ, and there are a number of ways in which traumatic damage occurs, ultimately resulting in degradation of articular cartilage. It was recognized in 1966 that articular cartilage change that accompanied osteochondral fragmentation could also be associated with concurrent traumatic damage to the attachment of the joint capsule and ligaments (Raker et al., 1966). However, there was little association made between primary disease in the synovial membrane and fibrous joint capsule and the development of osteoarthritic change in the articular cartilage until an experimental study demonstrated that cartilage degradation could occur in the horse in the absence of instability or trau- matic disruption of tissue and that loss of glycosaminoglycan (GAG) staining was associated with early morphologic breakdown at the surface of the cartilage (McIlwraith and Van Sickle, 1984). Surveys have confirmed that approximately 60% of lameness problems are related to osteoarthritis (National Animal Health Monitoring Systems, 2000; Caron and Genovese, 2003). Rapid resolution of synovitis and capsuli- tis is a critical part of the medical treatment of joint disease because of the principal role of synovitis in causing cartilage matrix breakdown. The goal of treatment of traumatic entities of the joint is twofold: (1) returning the joint to normal as quickly as possible, and (2) preventing the occurrence or reduction of the severity of osteoarthritis. In other words, treatment is intended to (1) reduce pain (lameness), and (2) minimize progression of joint deterioration. -

Pharmaceuticals As Environmental Contaminants
PharmaceuticalsPharmaceuticals asas EnvironmentalEnvironmental Contaminants:Contaminants: anan OverviewOverview ofof thethe ScienceScience Christian G. Daughton, Ph.D. Chief, Environmental Chemistry Branch Environmental Sciences Division National Exposure Research Laboratory Office of Research and Development Environmental Protection Agency Las Vegas, Nevada 89119 [email protected] Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada Why and how do drugs contaminate the environment? What might it all mean? How do we prevent it? Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada This talk presents only a cursory overview of some of the many science issues surrounding the topic of pharmaceuticals as environmental contaminants Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada A Clarification We sometimes loosely (but incorrectly) refer to drugs, medicines, medications, or pharmaceuticals as being the substances that contaminant the environment. The actual environmental contaminants, however, are the active pharmaceutical ingredients – APIs. These terms are all often used interchangeably Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada Office of Research and Development Available: http://www.epa.gov/nerlesd1/chemistry/pharma/image/drawing.pdfNational -

Pharmacotherapy for Interstitial Cystitis/Bladder Pain Syndrome
Current Bladder Dysfunction Reports (2019) 14:365–376 https://doi.org/10.1007/s11884-019-00540-9 PHARMACOTHERAPIES AND DRUG DEVELOPMENT/AGENTS (ES ROVNER, SECTION EDITOR) Pharmacotherapy for Interstitial Cystitis/Bladder Pain Syndrome Alyssa Greiman1 & Lindsey Cox 1 Published online: 6 November 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Purpose of Review Current literature regarding pharmacotherapy treatment strategies available for the management of interstitial cystitis/bladder pain syndrome (IC/BPS) will be addressed including oral, transdermal, and intravesical therapies. Pharmacotherapies with emerging data will be addressed, but the focus is on those treatments described by the AUA guidelines for IC/BPS. Recent Findings While multiple pharmacotherapy options for the management of IC/BPS exist, the evidence for most medical therapies is not strong and frequently yields mixed results. It has been over two decades since a new medication has gained FDA approval for the treatment of IC/BPS. This has prompted clinicians to reassess the approach to evaluating patients with IC/BPS, leading to the advent of phenotype-directed multimodal therapy. Summary Though national and international guidelines recommend a step-wise treatment algorithm beginning with the most conservative treatment options, the evidence for most therapies is mixed. Furthermore, recent randomized controlled trials of promising treatment options have yielded negative results, highlighting the importance of phenotype-directed classification to aid in the current management of IC/BPS and to allow for better research trial designs. Keywords Interstitial cystitis . Bladder pain syndrome . Pentosan polysulfate . Pharmacotherapy Introduction of infection or other identifiable causes.” [1]Thisisthe definition used by the American Urological Association Much of the difficulty surrounding treatment of interstitial (AUA),andassuch,isthedefinitionusedinthescope cystitis or bladder pain syndrome (IC/BPS) centers on the of this review. -

Cardiovascular System Drug Poster
Cardiovascular Drugs Created by the Njardarson Group (The University of Arizona): Edon Vitaku, Elizabeth A. Ilardi, Daniel J. Mack, Monica A. Fallon, Erik B. Gerlach, Miyant’e Y. Newton, Angela N. Yazzie, Jón T. Njarðarson Ethanol Glyceryl Trinitrate Anestisine Quinidine Procaine Adrenaline Adenosine Phenylepherine Heparin Vasoxyl Sotradecol Xylocaine Pronestyl Betamethasone Catapres Cedilanide Dopamine Inversine Metaraminol Acetyldigitoxin Hydrocortisone ( Ethanol ) ( Nitroglycerin ) ( Benzocaine ) ( Quinidine ) ( Procaine ) ( Epinephrine ) ( Adenosine ) ( Phenylephrine ) ( Heparin ) ( Methoxamine ) ( Sodium Tetradecyl Sulfate ) ( Lidocaine ) ( Procainamide ) ( Betamethasone ) ( Clonidine ) ( Deslanoside ) ( Dopamine ) ( Mecamylamine ) ( Metaraminol ) ( Acetyldigitoxin ) ( Hydrocortisone ) VASOPROTECTIVE CARDIAC THERAPY VASOPROTECTIVE CARDIAC THERAPY VASOPROTECTIVE CARDIAC THERAPY CARDIAC THERAPY CARDIAC THERAPY VASOPROTECTIVE CARDIAC THERAPY VASOPROTECTIVE CARDIAC THERAPY CARDIAC THERAPY VASOPROTECTIVE ANTIHYPERTENSIVE CARDIAC THERAPY CARDIAC THERAPY ANTIHYPERTENSIVE CARDIAC THERAPY CARDIAC THERAPY VASOPROTECTIVE Approved 1700s Approved 1879 Approved 1890s Approved 1900s Approved 1903 Approved 1920s Approved 1929 Approved 1930s Approved 1935 Approved 1940s Approved 1946 Approved 1949 Approved 1950 Approved 1950s Approved 1950s Approved 1950s Approved 1950s Approved 1950s Approved 1951 Approved 1952 Approved 1952 Regitine Phenoxybenzamine Serpasol Rescinnamine Diuril Harmonyl Naturetin Hydrochlorothiazide Hydrocortamate Fastin Ismel -

PHP Compound List
PHP Compound List Pet Health Pharmacy can compound prescriptions in a variety of strengths and dosage forms. In addition, flavors may be added to make medications more palatable. Included below is a sample of our formulations, but please note, we compound many other medications not included on this list. Oral Transdermal Cream / Liquids Capsules Gel Otic Powder Ointment Compounded Drug Acepromazine Maleate Acetazolamide Acetylsalicylic Acid Allopurinol Alprazolam Aluminum Hydroxide Amantadine HCL Amikacin Aminocaproic Acid + Vit & Min Aminophylline Amitriptyline HCL Amlodipine Amoxicillin Apomorphine HCL Ascorbic Acid Atenolol Azathioprine Azithromycin Benazepril HCL Betamethasone Dip. Bethanechol Chloride Budesonide Buprenorphine Availability of medications is subject to change. Oral Transdermal Cream / Liquids Capsules Gel Otic Powder Ointment Buspirone HCL Calcitriol Calcium Acetate Calcium Carbonate Calcium Edetate Disodium Cephalexin Chlorambucil Chloramphenicol Base Chloramphenicol/Betamethasone Dip./Tris EDTA Chloramphenicol/Betamethasone Dip. Chloramphenicol / Prednisolone / Tetracaine Chlorpheniramine Maleate Chlorpromazine HCL Cimetidine Ciprofloxacin / Betamethasone Dip Ciprofloxacin HCL Cisapride Clarithromycin Clemastine Fumarate Clindamycin Clindamycin / Itraconazole Clomipramine HCL Clopidogrel Bisulfate Clotrimazole Clotrimazole/Erythromycin Ethylsuccinate Clotrimazole/Hydrocortisone Co-Enzyme Q10 Colchicine Copper Sulfate Cyclophosphamide Cyclosporin Cyproheptadine HCL Availability of medications is subject to change. -

Deductible EPO (DEPO) Commercial (Self-Funded Plans) Formulary
2021 California Exclusive Provider Organization (EPO) / Deductible EPO (DEPO) Commercial (Self-Funded Plans) Formulary (List of Covered Drugs) PLEASE READ: THIS DOCUMENT CONTAINS INFORMATION ABOUT THE DRUGS WE COVER WHEN YOU PARTICIPATE IN A KAISER PERMANENTE EXCLUSIVE PROVIDER ORGANIZATION (EPO) OR DEDUCTIBLE EPO (DEPO) SELF-FUNDED PLAN. This prescription drug formulary was updated on 9/01/2021 and is effective as of September 7, 2021. This formulary document is subject to change and may vary depending on your health plan. It does not provide information regarding specific coverage, including specific exclusions, copays, or coinsurances. That information can be found by referring to your Summary Plan Description or other Plan documents. For more recent information about which drug formulary applies to your plan, visit kp.org/formulary or for questions about your prescription benefits contact the Customer Service number on your ID card. What is the Kaiser Permanente California EPO/DEPO Commercial Self-Funded Formulary? The California EPO/DEPO Commercial Self-Funded Formulary is a list of covered drugs chosen by a group of Kaiser Permanente doctors and pharmacists known as the Pharmacy and Therapeutics Committee. The Committee meets regularly to evaluate and select drugs that are safe and effective for our members. This Formulary meets the requirements outlined under state law, regulations, and guidance for commercial plans. What drugs are covered? The Kaiser Permanente formulary includes medically necessary brand, generic, and specialty drugs listed on the California Commercial Formulary, providing the prescription is filled at a Kaiser Permanente, or an affiliated pharmacy, and other plan rules are followed. If you are prescribed a drug on the California Commercial Formulary, that drug will be provided under the terms of your drug benefit. -

B2B Vigilant Drug Program Non-Essential Medication List
OptumRx Vigilant Drug Program Non-essential drug strategy Effective January 1, 2021 Thousands of medications are available today with even more entering the market — sometimes at a higher cost without additional value. OptumRx can help ensure that your pharmacy benefit dollars cover only the most clinically appropriate and affordable medications. Non-essential drug strategy* — Excludes select products that are available in the market but may not be approved by the U.S. Food and Drug Administration (FDA) or are deemed non-essential by the OptumRx Pharmacy & Therapeutics (P&T) Committee. These high-dollar products offer little clinical value versus other well-tested, FDA-approved agents. *Programs are also available for clients who prefer prior authorization rather than an exclusion strategy. Drug Name Drug Name 1ST MEDX-PATCH/ LIDOCAINE EXTERNAL adenosine (diagnostic) PATCH 4-0.0375-5-20 % ADRENALIN NASAL 7T GUMMY ES ADVANCED ALLERGY COLLECTION 7T LIDO ADVANCED BASE PLUS ACACIA SUBCUTANEOUS ADYPHREN ACCUCAINE ADYPHREN AMP ACESO AG ADYPHREN AMP II ACREMONIUM ADYPHREN II ACTICOAT 7 AGONEAZE ACTICOAT ABSORBENT AKOVAZ ACTICOAT ANTIMICROBIAL ALA-QUIN ACTICOAT EXTERNAL SHEET 16"X16" , ALBA-DERM 4"X48" , 4"X8" , 5"X5" , 8"X16" ALBUTEROL SULFATE INHALATION ACTICOAT FLEX 3 NEBULIZATION SOLUTION (5 MG/ML) 0.5% ACTICOAT FLEX 3 4"X4" alcohol injection ACTICOAT FLEX 7 ALCORTIN A ACTICOAT MOISTURE CONTROL ALDER EXTERNAL PAD 4"X4" , 4"X8" ALEVAMAX ACTICOAT SITE ALEVICYN ANTIPRURITIC ACTIVASE ALEVICYN ANTIPRURITIC SG EXTERNAL ACTIVE INJECTION BLM-1 GEL -

Managing Joint Disease in the Racehorse in the Face of Stricter Drug Restrictions
IN-DEPTH: RACING-RELATED LAMENESS Managing Joint Disease in the Racehorse in the Face of Stricter Drug Restrictions C. Wayne McIlwraith, BVSc, PhD, Diplomate ACVS, ACVSMR Author’s address: Gail Holmes Equine Orthopaedic Research Center, Department of Clinical Sciences, College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO 80523; e-mail: [email protected]. © 2013 AAEP. 1. Introduction racing to protect equine health and welfare. Par- Intra-articular use of corticosteroids has become a ticipants included analytical chemists, veterinary recent focus (or re-focus) of attention in the Thor- pharmacologists, veterinary surgeons, racing regu- oughbred racing industry. The clinical use and sci- latory veterinarians, and practicing racetrack veter- entific basis of intra-articular corticosteroid inarians. Among the recommendations was a administration including catastrophic injury, artic- prohibition on intra-articular use of corticosteroids ular cartilage degradation, and the development of within 7 days of a race, taking into consideration the osteoarthritis (OA) as well as the timing of injection concerns expressed by many participants about the relative to racing have been reviewed.1 Most re- proximity of intra-articular injections to race day. cently, there has been very specific examination by The experts also recommended a 72-hour with- the Racing, Medication, and Testing Consortium drawal time for dexamethasone, a commonly used (RMTC). At the time of that press release, the short-acting corticosteroid that can be administered RMTC had approved (1) minimal withdrawal time intravenously, intramuscularly and orally. Other recommendations for corticosteroids on the basis of short-acting corticosteroids would have similar re- both recently completed work funded in part by the strictions. -

Phase I Trial of Orally Administered Pentosan Polysulfate in Patients with Advanced Cancer1 in Vitroand
Vol. 3, 2347-2354. Decel?lber /997 Clinical Cancer Research 2347 Phase I Trial of Orally Administered Pentosan Polysulfate in Patients with Advanced Cancer1 John L. Marshall,2 Anton Welistein, James Rae, dose was increased, but their seventies were similar at all Robert J. DeLap, Kim Phipps, John Hanfelt, dose levels. There were no objective responses, although three patients had prolonged stabilization of previously pro- Manasses K. Yunmbam, Jim X. Sun, gressing disease. Pharmacokinetic analysis suggested Kenneth L. Duchin, and Michael J. Hawkins marked accumulation of PPS upon chronic administration. Lombardi Cancer Research Center, Georgetown University Hospital. Serum and urine bFGF levels failed to show a consistent, Washington. DC 2X)7 Ii. L. M.. A. W.. J. R.. K. P.. J. H.. M. J. H.J: Baker Norton Pharmaceuticals. Inc.. Miami. Florida 33137 Ii. X. S.. interpretable pattern; however the data suggested an in- K. L. D.j: Division of Oncology Drug Products. Center ftr Drug verse relationship between PPS and bFGF levels in s’ivo. A Evaluation and Research. Food and Drug Administration. Rockville. MTD could not be determined using the daily t.i.d. dosing Maryland 20854 lR. J. Dl: and NIH. National Cancer Institute. schedule due to the development of grade 3/4 GI toxicity Bethesda. Maryland 20892 [M. K. Y.] (proctitis) at all dose levels studied. PPS, administered p.o. at doses of 400 mg/m2 t.i.d., did not cause significant sys- ABSTRACT temic toxicity, but most patients developed moderate-to- Tumor angiogenesis is critically important to tumor severe GI toxicity within 1-2 months.