Short QT Syndrome: a Case Report and Review of Literatureଝ
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non Commercial Use Only
Cardiogenetics 2017; volume 7:6304 Sudden death in a young patient with atrial fibrillation Case Report Correspondence: María Angeles Espinosa Castro, Inherited Cardiovascular Disease A 22-year-old man suffered a sudden Program, Cardiology Department, Gregorio María Tamargo, cardiac arrest without previous symptoms Marañón Hospital, Dr. Esquerdo, 46, 28007, María Ángeles Espinosa, while he was at rest, waiting for a subway Madrid, Spain. Víctor Gómez-Carrillo, Miriam Juárez, train. Cardiopulmonary resuscitation was Tel.: +34.91.586.82.90. immediately started using an Automated E-mail: [email protected] Francisco Fernández-Avilés, External Defibrillation that identified the Raquel Yotti Key words: KCNQ1; mutation; channelopa- presence of ventricular fibrillation and thy; sudden cardiac death; atrial fibrillation. Inherited Cardiovascular Disease delivered a shock. Return of spontaneous Program, Cardiology Department, circulation was achieved after three Contributions: MT, acquisition and interpreta- Gregorio Marañón Hospital, Madrid, attempts, being atrial fibrillation (AF) the tion of data for the work, ensuring that ques- Spain patient’s rhythm at this point (Figure 1). tions related to the accuracy or integrity of any He was admitted to our Cardiovascular part of the work is appropriately investigated Intensive Care Unit and therapeutic and resolved; MAE, conception of the work, hypothermia was performed over a period critical revision of the intellectual content, final approval of the version to be published, Abstract of 24 h. After completing hypothermia, ensuring that questions related to the accuracy rewarming, and another 24 h of controlled of any part of the work is appropriately inves- Sudden cardiac death (SCD) in young normothermia the patient awakened with no tigated and resolved; VG-C, acquisition and patients without structural heart disease is residual neurologic damage. -
Atrial Arrhythmia Triggering Electromechanical Dissociation And
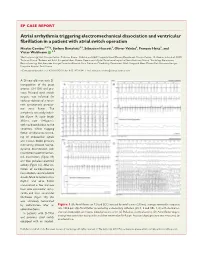
EP CASE REPORT ....................................................................................................................................................... Atrial arrhythmia triggering electromechanical dissociation and ventricular fibrillation in a patient with atrial switch operation Nicolas Combes1,2,3*, Stefano Bartoletti1,2,Se´bastien Hascoet€ 3, Olivier Vahdat2, Franc¸ois Heitz2, and Victor Waldmann 4,5 1Electrophysiology Unit, Clinique Pasteur, Toulouse, France; 2Pediatric and Adult Congenital Heart Disease Department, Clinique Pasteur, 45, Avenue de Lombez, 31076 Toulouse, France; 3Pediatric and Adult Congenital Heart Disease Department, Hoˆpital Marie Lannelongue, Le Plessis-Robinson, France; 4Cardiology Department, Electrophysiology Unit, European Georges Pompidou Hospital, Paris, France; and 5Cardiology Department, Adult Congenital Heart Disease Unit, European Georges Pompidou Hospital, Paris, France * Corresponding author. Tel: 133 562213131; fax: 133 562211641. E-mail address: [email protected] A 26-year-old man with D- transposition of the great arteries (D-TGA) and pre- vious Mustard atrial switch surgery was referred for catheter ablation of a recur- rent symptomatic paroxys- mal atrial flutter. The arrhythmia was easily induci- ble (Figure 1A, cycle length 310 ms, rate 194 b.p.m.), with rapid conduction to the ventricles. While mapping flutter, simultaneous record- ing of endocardial signals and invasive blood pressure monitoring showed haemo- dynamic deterioration with intermittent electromechan- ical dissociation (Figure 1B) and then pulseless electrical activity (Figure 1C). After ini- tiation of cardiopulmonary resuscitation, several electric shocks failed to restore sinus rhythm and atrial flutter transitioned a few minutes later into ventricular tachy- cardia and then ventricular fibrillation (Figure 1D); this was ultimately terminated by defibrillation after an Figure 1 (A) Atrial flutter on 12-lead ECG induced by atrial bursts (240 ms), average ventricular response adrenaline bolus. -

Mitral Valve Prolapse, Arrhythmias, and Sudden Cardiac Death: the Role of Multimodality Imaging to Detect High-Risk Features
diagnostics Review Mitral Valve Prolapse, Arrhythmias, and Sudden Cardiac Death: The Role of Multimodality Imaging to Detect High-Risk Features Anna Giulia Pavon 1,2,*, Pierre Monney 1,2,3 and Juerg Schwitter 1,2,3 1 Cardiac MR Center (CRMC), Lausanne University Hospital (CHUV), 1100 Lausanne, Switzerland; [email protected] (P.M.); [email protected] (J.S.) 2 Cardiovascular Department, Division of Cardiology, Lausanne University Hospital (CHUV), 1100 Lausanne, Switzerland 3 Faculty of Biology and Medicine, University of Lausanne (UniL), 1100 Lausanne, Switzerland * Correspondence: [email protected]; Tel.: +41-775-566-983 Abstract: Mitral valve prolapse (MVP) was first described in the 1960s, and it is usually a benign condition. However, a subtype of patients are known to have a higher incidence of ventricular arrhythmias and sudden cardiac death, the so called “arrhythmic MVP.” In recent years, several studies have been published to identify the most important clinical features to distinguish the benign form from the potentially lethal one in order to personalize patient’s treatment and follow-up. In this review, we specifically focused on red flags for increased arrhythmic risk to whom the cardiologist must be aware of while performing a cardiovascular imaging evaluation in patients with MVP. Keywords: mitral valve prolapse; arrhythmias; cardiovascular magnetic resonance Citation: Pavon, A.G.; Monney, P.; Schwitter, J. Mitral Valve Prolapse, Arrhythmias, and Sudden Cardiac Death: The Role of Multimodality 1. Mitral Valve and Arrhythmias: A Long Story Short Imaging to Detect High-Risk Features. In the recent years, the scientific community has begun to pay increasing attention Diagnostics 2021, 11, 683. -

Constrictive Pericarditis Causing Ventricular Tachycardia.Pdf
EP CASE REPORT ....................................................................................................................................................... A visually striking calcific band causing monomorphic ventricular tachycardia as a first presentation of constrictive pericarditis Kian Sabzevari 1*, Eva Sammut2, and Palash Barman1 1Bristol Heart Institute, UH Bristol NHS Trust UK, UK; and 2Bristol Heart Institute, UH Bristol NHS Trust UK & University of Bristol, UK * Corresponding author. Tel: 447794900287; fax: 441173425926. E-mail address: [email protected] Introduction Constrictive pericarditis (CP) is a rare condition caused by thickening and stiffening of the pericar- dium manifesting in dia- stolic dysfunction and enhanced interventricu- lar dependence. In the developed world, most cases are idiopathic or are associated with pre- vious cardiac surgery or irradiation. Tuberculosis remains a leading cause in developing areas.1 Most commonly, CP presents with symptoms of heart failure and chest discomfort. Atrial arrhythmias have been described as a rare pre- sentation, but arrhyth- mias of ventricular origin have not been reported. Figure 1 (A) The 12 lead electrocardiogram during sustained ventricular tachycardia is shown; (B and C) Case report Different projections of three-dimensional reconstructions of cardiac computed tomography demonstrating a A 49-year-old man with a striking band of calcification around the annulus; (D) Carto 3DVR mapping—the left hand panel (i) demonstrates a background of diabetes, sinus beat with late potentials at the point of ablation in the coronary sinus, the right hand panel (iii) shows the hypertension, and hyper- pacemap with a 89% match to the clinical tachycardia [matching the morphology seen on 12 lead ECG (A)], and cholesterolaemia and a the middle panel (ii) displays the three-dimensional voltage map. -

Cardiac Arrest and Ventricular Fibrillation * a Method of Treatment by Electrical Shock
Thorax: first published as 10.1136/thx.7.3.205 on 1 September 1952. Downloaded from Thorax (1952), 7, 205. CARDIAC ARREST AND VENTRICULAR FIBRILLATION * A METHOD OF TREATMENT BY ELECTRICAL SHOCK BY I. K. R. MCMILLANt F. B. COCKETT, AND P. STYLES Froml the Departnment of Cardiology, tile Professorial Surgical Unit, and tile Departalzent of Phlysical Medicine, St. Thomlas's Hospital, London (RECEIVED FOR PUBLICATION APRIL 30, 1952) Cardiac arrest during operation is a subject of VENTRICULAR FIBRILLATION importance to all surgeons. During intra-thoracic The treatment of ventricular fibrillation bv operations cardiac arrest and ventricular fibrilla- electrical shock therapy is not new and was first tion can be differentiated by direct observation of tried experimentally in 1899 (Prevost and Battelli). the heart. In recent years this method has been extensively Ventricular fibrillation is an incoordinated type investigated in the United States and France of contraction which produces no useful beats. (Kouwenhoven and Kay, 1951; Johnson and The typical rippling movements of the ventricular Kirby, 1951 ; Santy and Marion, 1950). muscle are unmistakable when seen or felt with The treatment of ventricular fibrillation by the hand directly on the heart. The treatment of zlectrical shock gives encouraging results in the copyright. a heart that has stopped is cardiac massage and experimental animal, and may be a useful adjunct adequate oxygenation. The treatment of ventri- in the operating theatre during cardiac and general cular fibrillation which we recommend is, first, surgery. Under these conditions shock therapy massage to maintain the oxygen supply to the can be rapidly applied. Provided that adequate heart through the coronary arteries, followed oxygenation is maintained and direct cardiac mas- http://thorax.bmj.com/ rapidly by electrical shocking to restore normal sage promptly initiated and maintained, shock rhythm therapy can be applied without undue haste. -

Effects of Nifekalant Injected Into the Pericardial Space on the Transmural Dispersion of Repolarization in Pig
Showa Univ J Med Sci 19(3), 123 135, September 2007 Original Effects of Nifekalant Injected into the Pericardial Space on the Transmural Dispersion of Repolarization in Pig Hiroyuki ITo, Taku ASANO, Youichi KOBAYASHI, Tatsuya ONUKI, Fumito MwosHI, Taka-aki MATSUYAMA, Yoshino MINOURA, Norikazu WATANABE, Mitsuharu KAWAMURA, Kaoru TANNO and Takashl KATAGIRI Abstract: Transmural dispersion of repolarization (TDR) has been implicated in the onset of ventricular arrhythmia. We investigated the effects of nifekalant (NIF) injected into the pericardial space on TDR and T wave in pig. We injected 50 or 100 mg of NIF into the pericardial space of 11 pigs, and measured the effective refractory period (ERP) between the endocardial and epicardial myocardial cells, as well as QT time and QT peak-end as an index of TDR and T waveforms, respectively. TDR decreased from 56•} 10 msec to 44•}8 msec (P < 0.01), 5.1 min after injection of 100 mg of NIF, although the QTc did not change. At a later time, QTc increased from 457 •} 44 msec before injection to 540•}49 msec (P < 0.01) and TDR recovered to the control level. When 50 mg of NIF was injected, the T wave amplitude decreased from 0.433 •}0.301 mV before injection to 0.107•}0.192 mV (P < 0.01) at 10 min after injection ; 100 mg of NIF caused the T wave amplitude to decrease more rapidly, reaching a negative value at 4.5 min after injection. Injecting NIF into the pericardial space altered ERP, QT, and T waveform, and also decreased TDR. -

What Are Premature Ventricular Contractions?
What Are Premature Ventricular Contractions? Premature ventricular contractions (VPCs or PVCs) are irregular beats that originate from the heart’s pumping chambers (ventricles). These beats interrupt the normal regular (sinus) rhythm, and result in irregularity to the heart rhythm. Although single PVCs are not life- threatening, they may indicate the presence of underlying heart disease, or when they become more severe can cause rapid and dangerous increases in heart rate (ventricular tachycardia). WHAT CAUSES PREMATURE VENTRICULAR CONTRACTIONS? PVCs can occur secondary to a variety of causes, most concerning being cardiac disease. PVCs are most commonly seen in dogs with Dilated Cardiomyopathy, but can also occur in the later stages of disease with Myxomatous Mitral Valve Degeneration. PVCs and other ventricular arrhythmias can also with no evidence of underlying structural heart disease. The workup for PVCs can be frustrating, because PVCs can also occur secondary to conditions unrelated to the heart. Most commonly they can occur secondary to gastrointestinal disease, systemic disease, and pain. HOW ARE PREMATURE VENTRICULAR CONTRACTIONS DIAGNOSED? Most commonly, your veterinarian will hear an irregular heart rhythm and recommend an electrocardiogram (ECG) to assess the heart rhythm. The ECG will enable your veterinarian to determine whether the irregular rhythm is due to PVCs. Following this diagnosis, there are several additional recommended tests. TESTING Following this diagnosis, there are several additional recommended tests. ECHOCARDIOGRAPHY Echocardiography (heart ultrasound) is recommended to make sure that there is no evidence of structural cardiac disease causing the abnormal heart rhythm. 24-HOUR HOLTER Single PVCs usually do not require treatment, however a Holter monitor will determine whether the frequency or severity of the MONITOR PVCs warrant anti-arrhythmic medication, and make sure that there is no evidence of ventricular tachycardia, which is a life threatening heart rhythm. -

The Refractory VF Arrest Patient: a Review of the Current Treatment
1/24/2018 The Refractory VF Arrest Patient: A Review of the Current Treatment Options Marc Conterato, MD, FACEP Office of the Medical Director North Memorial Health Ambulance Service DISCLOSURE STATEMENT • Board Member, MN Resuscitation Consortium - Images of any commercial devices or medications are for illustration purposes only. The inclusion of such images in this presentation does not imply endorsement of any specific device or company. 2 Objectives • Delineate Refractory Ventricular Fibrillation (RVF) • Recurrent VF versus Refractory VF • What is “Electrical Storm” • Current Pre-hospital treatments • Additional hospital treatments • Dual/Double Sequential Defibrillation • ECMO/ECLS-A New Hope? 3 1 1/24/2018 A Standard Scenario • 55 YOF collapses at stop light, and her car rolls into the car in front of her. Airbags do not deploy, and bystanders find her slumped over the steering wheel and pulseless. • First responder/bystanders start CPR, and deliver three AED shocks prior to EMS arrival. • On EMS arrival, a fourth shock is delivered, an IO and alternative airway placed. Automated CPR started. • Epinephrine 2 mg (total) and Amiodarone 300 mg given, and the patient remains in VF. • Patient downtime is now ~25 minutes, and repeat evaluation reveals persistent VF. 4 What are your options • Continue CPR for a total of 30 minutes with recurrent defibrillations, additional epinephrine, bicarbonate and Amiodarone? • “Load and Go” to the local hospital with CPR enroute and continuing the resuscitation? • “Load and Go” to the local CCL (Cardiac Cath Lab) hospital with CPR enroute and continuing the resuscitation, with possible PCI with ongoing CPR? • Call HEMS unit for transfer to CCL hospital, but can they continue effective CPR in the helicopter? • “Load and Go” to a ECMO/ECLS center with CPR enroute and continuing the resuscitation, on the basis they can accept the patient and continue resuscitation? 5 Defining the problem: What is recurrent versus refractory VF (RVF)? • Recurrent VF is a rhythm that terminates with cardioversion, but then recurs rapidly. -

Basic Rhythm Recognition
Electrocardiographic Interpretation Basic Rhythm Recognition William Brady, MD Department of Emergency Medicine Cardiac Rhythms Anatomy of a Rhythm Strip A Review of the Electrical System Intrinsic Pacemakers Cells These cells have property known as “Automaticity”— means they can spontaneously depolarize. Sinus Node Primary pacemaker Fires at a rate of 60-100 bpm AV Junction Fires at a rate of 40-60 bpm Ventricular (Purkinje Fibers) Less than 40 bpm What’s Normal P Wave Atrial Depolarization PR Interval (Normal 0.12-0.20) Beginning of the P to onset of QRS QRS Ventricular Depolarization QRS Interval (Normal <0.10) Period (or length of time) it takes for the ventricles to depolarize The Key to Success… …A systematic approach! Rate Rhythm P Waves PR Interval P and QRS Correlation QRS Rate Pacemaker A rather ill patient……… Very apparent inferolateral STEMI……with less apparent complete heart block RATE . Fast vs Slow . QRS Width Narrow QRS Wide QRS Narrow QRS Wide QRS Tachycardia Tachycardia Bradycardia Bradycardia Regular Irregular Regular Irregular Sinus Brady Idioventricular A-Fib / Flutter Bradycardia w/ BBB Sinus Tach A-Fib VT PVT Junctional 2 AVB / II PSVT A-Flutter SVT aberrant A-Fib 1 AVB 3 AVB A-Flutter MAT 2 AVB / I or II PAT PAT 3 AVB ST PAC / PVC Stability Hypotension / hypoperfusion Altered mental status Chest pain – Coronary ischemic Dyspnea – Pulmonary edema Sinus Rhythm Sinus Rhythm P Wave PR Interval QRS Rate Rhythm Pacemaker Comment . Before . Constant, . Rate 60-100 . Regular . SA Node Upright in each QRS regular . Interval =/< leads I, II, . Look . Interval .12- .10 & III alike .20 Conduction Image reference: Cardionetics/ http://www.cardionetics.com/docs/healthcr/ecg/arrhy/0100_bd.htm Sinus Pause A delay of activation within the atria for a period between 1.7 and 3 seconds A palpitation is likely to be felt by the patient as the sinus beat following the pause may be a heavy beat. -

PAROXYSMAL VENTRICULAR TACHYCARDIA OCCURRING in a NORMAL HEART by DAVID ROMNEY, M.B., B.Ch.(Dub.) Ex-Senior House Officer in Medicine, St
Postgrad Med J: first published as 10.1136/pgmj.31.354.191 on 1 April 1955. Downloaded from I9I -J PAROXYSMAL VENTRICULAR TACHYCARDIA OCCURRING IN A NORMAL HEART By DAVID ROMNEY, M.B., B.Ch.(Dub.) Ex-Senior House Officer in Medicine, St. James's Hospital, Balham It is widely taught-and rightly so-that by sinus pressure will, of course, not affect the paroxysmal ventricular tachycardia is one of the rate in ventricular tachycardia. rarer arrhythmias, and is associated with grave In I953 Froment, Gallavardin and Cahen myocardial damage, with special reference to myo- offered a classification of various forms of cardial infarction. It is the least common, and paroxysmal ventricular tachycardia, and included most serious of the paroxysmal tachycardias, some case report. They described the following which contain four varieties of arrhythmia: supra- groups:- ventricular (auricular and nodal) 60 per cent.; (i) Terminal prefibrillatory ventricular tachy- auricular fibrillation, 30 per cent.; auricular flutter, cardia. 6 per cent.; and ventricular tachycardia, 4 per (ii) Curable and mild monomorphic extra- cent. systoles, with paroxysms of tachycardia. Figures from various sources (Campbell, 1947) (iii) Paroxysmal ventricular tachycardia due toby copyright. would indicate that in the latter group, four-fifths a lesion of the ventricular septum. of the cases had seriously damaged hearts. In (iv) Persistent and prolonged ventricular tachy- the remaining fifth no cause was found for the cardia developing in sound hearts, usually paroxysms. Paroxysmal ventricular tachycardia is in young subjects. more common in men than in women in the proportion of about 3.2. This arrhythmia was Case Report first identified by Sir Thomas Lewis in I909 and A married woman, aged 48, first became aware Gallavardin (1920, I921, 1922) emphasized the in 1948 of a paroxysm which caused alarm, faint- seriousness of the 'terminal ven- ness and It lasted a short pre-fibrillatory collapse. -

Pub 100-04 Medicare Claims Processing Centers for Medicare & Medicaid Services (CMS) Transmittal 3054 Date: August 29, 2014 Change Request 8803
Department of Health & CMS Manual System Human Services (DHHS) Pub 100-04 Medicare Claims Processing Centers for Medicare & Medicaid Services (CMS) Transmittal 3054 Date: August 29, 2014 Change Request 8803 SUBJECT: Ventricular Assist Devices for Bridge-to-Transplant and Destination Therapy I. SUMMARY OF CHANGES: This Change Request (CR) is effective for claims with dates of service on and after October 30, 2013; contractors shall pay claims for Ventricular Assist Devices as destination therapy using the criteria in Pub. 100-03, part 1, section 20.9.1, and Pub. 100-04, Chapter 32, sec. 320. EFFECTIVE DATE: October 30, 2013 *Unless otherwise specified, the effective date is the date of service. IMPLEMENTATION DATE: September 30, 2014 Disclaimer for manual changes only: The revision date and transmittal number apply only to red italicized material. Any other material was previously published and remains unchanged. However, if this revision contains a table of contents, you will receive the new/revised information only, and not the entire table of contents. II. CHANGES IN MANUAL INSTRUCTIONS: (N/A if manual is not updated) R=REVISED, N=NEW, D=DELETED-Only One Per Row. R/N/D CHAPTER / SECTION / SUBSECTION / TITLE D 3/90.2.1/Artifiical Hearts and Related Devices R 32/Table of Contents N 32/320/Artificial Hearts and Related Devices N 32/320.1/Coding Requirements for Furnished Before May 1, 2008 N 32/320.2/Coding Requirements for Furnished After May 1, 2008 N 32/320.3/ Ventricular Assist Devices N 32/320.3.1/Postcardiotomy N 32/320.3.2/Bridge-To -Transplantation (BTT) N 32/320.3.3/Destination Therapy (DT) N 32/320.3.4/ Other N 32/320.4/ Replacement Accessories and Supplies for External Ventricular Assist Devices or Any Ventricular Assist Device (VAD) III. -

Genetic Testing for Hereditary Cardiac Disease
Clinical Appropriateness Guidelines Genetic Testing for Hereditary Cardiac Disease EFFECTIVE MARCH 8, 2021 Appropriate.Safe.Affordable © 2019 AIM Specialty Health 2064-0319 Table of Contents Scope .......................................................................................................................................................... 3 Genetic Counseling Requirement ............................................................................................................... 3 Appropriate Use Criteria.............................................................................................................................. 3 Confirmation/Diagnostic Testing of Affected Individuals .............................................................................. 4 Testing of Asymptomatic Individuals .............................................................................................................. 4 Post-Mortem Testing ........................................................................................................................................ 4 Long QT ............................................................................................................................................................. 5 Dilated Cardiomyopathy .................................................................................................................................. 5 Tests Not Clinically Appropriate .....................................................................................................................