Combating Malignant Melanoma with the Multifaceted Soy-Dervied Peptide Lunasin." (2017)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The B Vitamins Nicotinamide (B3) and Riboflavin (B2)
The B Vitamins Nicotinamide (B3) and Riboflavin (B2) Stimulate Metamorphosis in Larvae of the Deposit- Feeding Polychaete Capitella teleta: Implications for a Sensory Ligand-Gated Ion Channel Robert T. Burns1*, Jan A. Pechenik1, William J. Biggers2, Gia Scavo2, Christopher Lehman2 1 Department of Biology, Tufts University, Medford, Massachusetts, United States of America, 2 Department of Biology, Wilkes University, Wilkes-Barre, Pennsylvania, United States of America Abstract Marine sediments can contain B vitamins, presumably incorporated from settled, decaying phytoplankton and microorganisms associated with decomposition. Because B vitamins may be advantageous for the energetically intensive processes of metamorphosis, post-metamorphic growth, and reproduction, we tested several B vitamins to determine if they would stimulate larvae of the deposit-feeding polychaete Capitella teleta to settle and metamorphose. Nicotinamide and riboflavin individually stimulated larvae of C. teleta to settle and metamorphose, generally within 1–2 hours at nicotinamide concentrations as low as 3 mM and riboflavin concentrations as low as 50 mM. More than 80% of the larvae metamorphosed within 30 minutes at a nicotinamide concentration of 7 mM. The pyridine channel agonist pyrazinecarboxamide also stimulated metamorphosis at very low concentrations. In contrast, neither lumichrome, thiamine HCl, pyridoxine HCl, nor vitamin B12 stimulated larvae of C. teleta to metamorphose at concentrations as high as 500 mM. Larvae also did not metamorphose in response to either nicotinamide or pyrazinecarboxamide in calcium-free seawater or with the addition of 4-acetylpyridine, a competitive inhibitor of the pyridine receptor. Together, these results suggest that larvae of C. teleta are responding to nicotinamide and riboflavin via a chemosensory pyridine receptor similar to that previously reported to be present on crayfish chela and involved with food recognition. -

Neural Stem Cell-Derived Exosomes Revert HFD-Dependent Memory Impairment Via CREB-BDNF Signalling
International Journal of Molecular Sciences Article Neural Stem Cell-Derived Exosomes Revert HFD-Dependent Memory Impairment via CREB-BDNF Signalling Matteo Spinelli 1, Francesca Natale 1,2, Marco Rinaudo 1, Lucia Leone 1,2, Daniele Mezzogori 1, Salvatore Fusco 1,2,* and Claudio Grassi 1,2 1 Department of Neuroscience, Università Cattolica del Sacro Cuore, 00168 Rome, Italy; [email protected] (M.S.); [email protected] (F.N.); [email protected] (M.R.); [email protected] (L.L.); [email protected] (D.M.); [email protected] (C.G.) 2 Fondazione Policlinico Universitario Agostino Gemelli IRCCS, 00168 Rome, Italy * Correspondence: [email protected] Received: 6 October 2020; Accepted: 25 November 2020; Published: 26 November 2020 Abstract: Overnutrition and metabolic disorders impair cognitive functions through molecular mechanisms still poorly understood. In mice fed with a high fat diet (HFD) we analysed the expression of synaptic plasticity-related genes and the activation of cAMP response element-binding protein (CREB)-brain-derived neurotrophic factor (BDNF)-tropomyosin receptor kinase B (TrkB) signalling. We found that a HFD inhibited both CREB phosphorylation and the expression of a set of CREB target genes in the hippocampus. The intranasal administration of neural stem cell (NSC)-derived exosomes (exo-NSC) epigenetically restored the transcription of Bdnf, nNOS, Sirt1, Egr3, and RelA genes by inducing the recruitment of CREB on their regulatory sequences. Finally, exo-NSC administration rescued both BDNF signalling and memory in HFD mice. Collectively, our findings highlight novel mechanisms underlying HFD-related memory impairment and provide evidence of the potential therapeutic effect of exo-NSC against metabolic disease-related cognitive decline. -

Oup Radres Rrz001 289..297 ++
Journal of Radiation Research, Vol. 60, No. 3, 2019, pp. 289–297 doi: 10.1093/jrr/rrz001 Advance Access Publication: 26 February 2019 Ionizing radiation affects the composition of the proteome of extracellular vesicles released by head-and-neck cancer cells in vitro Agata Abramowicz1, Anna Wojakowska1, Lukasz Marczak2, Malgorzata Lysek-Gladysinska3, Mateusz Smolarz1, Michael D. Story4, Joanna Polanska5, Piotr Widlak1 and Monika Pietrowska1,* 1Center for Translational Research and Molecular Biology of Cancer, Maria Sklodowska–Curie Institute–Oncology Center, Gliwice Branch, ul. Wybrzeze Armii Krajowej 15, 44-101 Gliwice, Poland 2Institute of Bioorganic Chemistry, Polish Academy of Sciences, ul. Noskowskiego 12/14, 61-704 Poznan, Poland 3The Jan Kochanowski University in Kielce, Institute of Biology, Department of Cell Biology and Electron Microscopy, ul. Swietokrzyska 15, 25-406 Kielce, Poland 4University of Texas Southwestern Medical Center, Department of Radiation Oncology, Division of Molecular Radiation Biology, 5323 Harry Hines Boulevard, Dallas, TX 75390, USA 5Faculty of Automatic Control, Electronics and Computer Science, Silesian University of Technology, ul. Akademicka 16, 44-100 Gliwice, Poland *Corresponding author. Center for Translational Research and Molecular Biology of Cancer, Maria Sklodowska–Curie Institute–Oncology Center, Gliwice Branch, ul. Wybrzeze Armii Krajowej 15, 44-101 Gliwice, Poland. Tel: +0048-32-278-9627; Fax: +0048-32-278-9840; Email: [email protected] (Received 29 August 2018; revised 7 November 2018; editorial decision 8 January 2019) ABSTRACT Exosomes and other extracellular vesicles are key players in cell-to-cell communication, and it has been proposed that they are involved in different aspects of the response to ionizing radiation, including transmitting the radiation-induced bystander effect and mediating radioresistance. -

Neurotransmitters-Drugs Andbrain Function.Pdf
Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Edited by R. A. Webster Department of Pharmacology, University College London, UK JOHN WILEY & SONS, LTD Chichester Á New York Á Weinheim Á Brisbane Á Singapore Á Toronto Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Copyright # 2001 by John Wiley & Sons Ltd. Bans Lane, Chichester, West Sussex PO19 1UD, UK National 01243 779777 International ++44) 1243 779777 e-mail +for orders and customer service enquiries): [email protected] Visit our Home Page on: http://www.wiley.co.uk or http://www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1P0LP,UK, without the permission in writing of the publisher. Other Wiley Editorial Oces John Wiley & Sons, Inc., 605 Third Avenue, New York, NY 10158-0012, USA WILEY-VCH Verlag GmbH, Pappelallee 3, D-69469 Weinheim, Germany John Wiley & Sons Australia, Ltd. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supporting Information
Supporting Information Figure S1. The functionality of the tagged Arp6 and Swr1 was confirmed by monitoring cell growth and sensitivity to hydeoxyurea (HU). Five-fold serial dilutions of each strain were plated on YPD with or without 50 mM HU and incubated at 30°C or 37°C for 3 days. Figure S2. Localization of Arp6 and Swr1 on chromosome 3. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 3L, Tel 3R, CEN3, and the RP gene are shown under the panels. Figure S3. Localization of Arp6 and Swr1 on chromosome 4. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) in the whole chromosome region are compared. The position of Tel 4L, Tel 4R, CEN4, SWR1, and RP genes are shown under the panels. Figure S4. Localization of Arp6 and Swr1 on the region including the SWR1 gene of chromosome 4. The binding of Arp6- FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position and orientation of the SWR1 gene is shown. Figure S5. Localization of Arp6 and Swr1 on chromosome 5. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 5L, Tel 5R, CEN5, and the RP genes are shown under the panels. Figure S6. Preferential localization of Arp6 and Swr1 in the 5′ end of genes. Vertical bars represent the binding ratio of proteins in each locus. -

Investigation of Candidate Genes and Mechanisms Underlying Obesity
Prashanth et al. BMC Endocrine Disorders (2021) 21:80 https://doi.org/10.1186/s12902-021-00718-5 RESEARCH ARTICLE Open Access Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules G. Prashanth1 , Basavaraj Vastrad2 , Anandkumar Tengli3 , Chanabasayya Vastrad4* and Iranna Kotturshetti5 Abstract Background: Obesity associated type 2 diabetes mellitus is a metabolic disorder ; however, the etiology of obesity associated type 2 diabetes mellitus remains largely unknown. There is an urgent need to further broaden the understanding of the molecular mechanism associated in obesity associated type 2 diabetes mellitus. Methods: To screen the differentially expressed genes (DEGs) that might play essential roles in obesity associated type 2 diabetes mellitus, the publicly available expression profiling by high throughput sequencing data (GSE143319) was downloaded and screened for DEGs. Then, Gene Ontology (GO) and REACTOME pathway enrichment analysis were performed. The protein - protein interaction network, miRNA - target genes regulatory network and TF-target gene regulatory network were constructed and analyzed for identification of hub and target genes. The hub genes were validated by receiver operating characteristic (ROC) curve analysis and RT- PCR analysis. Finally, a molecular docking study was performed on over expressed proteins to predict the target small drug molecules. Results: A total of 820 DEGs were identified between -

Beyond Desensitization: Physiological Relevance of Arrestin-Dependent Signaling
0031-6997/10/6202-305–330$20.00 PHARMACOLOGICAL REVIEWS Vol. 62, No. 2 U.S. Government work not protected by U.S. copyright 2436/3586110 Pharmacol Rev 62:305–330, 2010 Printed in U.S.A. ASSOCIATE EDITOR: DAVID R. SIBLEY Beyond Desensitization: Physiological Relevance of Arrestin-Dependent Signaling LOUIS M. LUTTRELL AND DIANE GESTY-PALMER Departments of Medicine and Biochemistry & Molecular Biology, Medical University of South Carolina (L.M.L.) and the Ralph H. Johnson Veterans Affairs Medical Center (L.M.L.), Charleston, South Carolina; and Department of Medicine, Duke University Medical Center (D.G.-P.) and the Durham Veterans Affairs Medical Center (D.G.-P.), Durham, North Carolina Abstract ................................................................................ 305 I. Introduction............................................................................. 306 II. The duality of arrestin function ........................................................... 307 A. Arrestin-dependent desensitization and endocytosis...................................... 307 B. Arrestins as signal transducers ........................................................ 308 III. Arrestin signaling in vitro ................................................................ 310 A. Diversity in arrestin signaling ......................................................... 310 B. Receptor desensitization and second messenger production ............................... 311 C. Receptor endocytosis and vesicle trafficking ............................................ -

Understanding Allergic Multimorbidity Within the Non-Eosinophilic
University of Groningen Understanding allergic multimorbidity within the non-eosinophilic interactome Aguilar, Daniel; Lemonnier, Nathanael; Koppelman, Gerard H; Melén, Erik; Oliva, Baldo; Pinart, Mariona; Guerra, Stefano; Bousquet, Jean; Anto, Josep M Published in: PLoS ONE DOI: 10.1371/journal.pone.0224448 IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2019 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Aguilar, D., Lemonnier, N., Koppelman, G. H., Melén, E., Oliva, B., Pinart, M., Guerra, S., Bousquet, J., & Anto, J. M. (2019). Understanding allergic multimorbidity within the non-eosinophilic interactome. PLoS ONE, 14(11), [e0224448]. https://doi.org/10.1371/journal.pone.0224448 Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 26-12-2020 RESEARCH ARTICLE Understanding allergic multimorbidity within the non-eosinophilic interactome 1,2,3 4 5,6 7 Daniel AguilarID *, Nathanael LemonnierID , Gerard H. -

Ncounter® Mouse Autoimmune Profiling Panel - Gene and Probe Details
nCounter® Mouse AutoImmune Profiling Panel - Gene and Probe Details Official Symbol Accession Alias / Previous Symbol Official Full Name Other targets or Isoform Information AW208573,CD143,expressed sequence AW208573,MGD-MRK- Ace NM_009598.1 1032,MGI:2144508 angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 2610036I19Rik,2610510L13Rik,Acinus,apoptotic chromatin condensation inducer in the nucleus,C79325,expressed sequence C79325,MGI:1913562,MGI:1919776,MGI:2145862,mKIAA0670,RIKEN cDNA Acin1 NM_001085472.2 2610036I19 gene,RIKEN cDNA 2610510L13 gene apoptotic chromatin condensation inducer 1 Acp5 NM_001102405.1 MGD-MRK-1052,TRACP,TRAP acid phosphatase 5, tartrate resistant 2310066K23Rik,AA960180,AI851923,Arp1b,expressed sequence AA960180,expressed sequence AI851923,MGI:2138136,MGI:2138359,RIKEN Actr1b NM_146107.2 cDNA 2310066K23 gene ARP1 actin-related protein 1B, centractin beta Adam17 NM_001277266.1 CD156b,Tace,tumor necrosis factor-alpha converting enzyme a disintegrin and metallopeptidase domain 17 ADAR1,Adar1p110,Adar1p150,AV242451,expressed sequence Adar NM_001038587.3 AV242451,MGI:2139942,mZaADAR adenosine deaminase, RNA-specific Adora2a NM_009630.2 A2AAR,A2aR,A2a, Rs,AA2AR,MGD-MRK-16163 adenosine A2a receptor Ager NM_007425.2 RAGE advanced glycosylation end product-specific receptor AI265500,angiotensin precursor,Aogen,expressed sequence AI265500,MGD- Agt NM_007428.3 MRK-1192,MGI:2142488,Serpina8 angiotensinogen (serpin peptidase inhibitor, clade A, member 8) Ah,Ahh,Ahre,aromatic hydrocarbon responsiveness,aryl hydrocarbon -
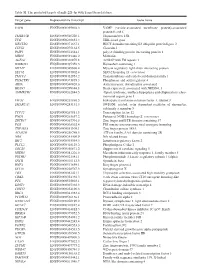
Table SI. the Predicted Targets of Mir-221-3P with Targetscan Database
Table SI. The predicted targets of miR-221-3p with TargetScan database. Target gene Representative transcript Gene name VAPB ENST00000395802.3 VAMP (vesicle-associated membrane protein)-associated protein B and C TMSB15B ENST00000540220.1 Thymosin beta 15B TFG ENST00000240851.4 TRK-fused gene HECTD2 ENST00000371667.1 HECT domain containing E3 ubiquitin protein ligase 2 CLVS2 ENST00000275162.5 Clavesin 2 PAIP1 ENST00000514514.1 poly(A) binding protein interacting protein 1 MIDN ENST00000591446.2 Midnolin AGFG1 ENST00000310078.8 ArfGAP with FG repeats 1 HMBOX1 ENST00000397358.3 Homeobox containing 1 MYLIP ENST00000349606.4 Myosin regulatory light chain interacting protein SEC62 ENST00000337002.4 SEC62 homolog (S. cerevisiae) TMCC1 ENST00000432054.2 Transmembrane and coiled-coil domain family 1 PHACTR4 ENST00000373839.3 Phosphatase and actin regulator 4 AIDA ENST00000340020.6 Axin interactor, dorsalization associated BEAN1 ENST00000299694.8 Brain expressed, associated with NEDD4, 1 AMMECR1 ENST00000262844.5 Alport syndrome, midface hypoplasia and elliptocytosis chro- mosomal region gene 1 EIF3J ENST00000261868.5 Eukaryotic translation initiation factor 3, subunit J SMARCA5 ENST00000283131.3 SWI/SNF related, actin dependent regulator of chromatin, subfamily a, member 5 TCF12 ENST00000267811.5 Transcription factor 12 PNO1 ENST00000263657.2 Partner of NOB1 homolog (S. cerevisiae) ZBTB37 ENST00000367701.5 Zinc finger and BTB domain containing 37 FOS ENST00000303562.4 FBJ murine osteosarcoma viral oncogene homolog ZNF385A ENST00000551109.1 Zinc -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. The CX3CR1/CX3CL1 Axis Drives the Migration and Maturation of Oligodendroglia in the Central Nervous System Catriona Ford Thesis Submitted for the Degree of Doctor of Philosophy The University of Edinburgh 2017 Abstract In the central nervous system, the axons of neurons are protected from damage and aided in electrical conductivity by the myelin sheath, a complex of proteins and lipids formed by oligodendrocytes. Loss or damage to the myelin sheath may result in impairment of electrical axonal conduction and eventually to neuronal death. Such demyelination is responsible, at least in part, for the disabling neurodegeneration observed in pathologies such as Multiple Sclerosis (MS) and Spinal Cord Injury. In the regenerative process of remyelination, oligodendrocyte precursor cells (OPCs), the resident glial stem cell population of the adult CNS, migrate toward the injury site, proliferate and differentiate into adult oligodendrocytes which subsequently reform the myelin sheath.