Supplemental Figures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Associations of Serotonin Receptor Gene HTR3A, HTR3B, and HTR3A Haplotypes with Bipolar Disorder in Chinese Patients
Associations of serotonin receptor gene HTR3A, HTR3B, and HTR3A haplotypes with bipolar disorder in Chinese patients J. Jian1,2*, C. Li1,2*, J. Xu3, D. Qiao2, G. Mi2, X. Chen2 and M. Tang2 1Shandong University School of Medicine, Ji’nan, Shandong, China 2Shandong Mental Health Center Ji’nan, Shandong, China 3Business Management Department, Shandong Center for Disease Control and Prevention, Jinan, Shandong, China *These authors contributed equally to this study. Corresponding author: M. Tang E-mail: [email protected] Genet. Mol. Res. 15 (3): gmr.15038671 Received March 30, 2016 Accepted August 8, 2016 Published September 16, 2016 DOI http://dx.doi.org/10.4238/gmr.15038671 Copyright © 2016 The Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution ShareAlike (CC BY-SA) 4.0 License. ABSTRACT. Single nucleotide polymorphisms (SNPs) in HTR3A and HTR3B have been reported to be associated with bipolar disorder in European and Japanese populations. We explored the roles of 21 tag SNPs in HTR3A and HTR3B in susceptibility to bipolar disorder in a Chinese cohort. Twenty-one Tag SNPs were genotyped in a study consisting of 130 patients with bipolar disorder, who visited Shandong Mental Health Center between June 2013 and May 2014, and 109 healthy individuals as controls. All of the tag SNPs were genotyped using Sequenom MassArray matrix-assisted laser desorption/ionization time of flight spectrometry. Plink 1.07, Haploview 4.2, and SPSS 20.0 were used for the analysis of the genotypes and the associations of the haplotypes with bipolar disorder. Association analyses of tag Genetics and Molecular Research 15 (3): gmr.15038671 J. -
![Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]](https://docslib.b-cdn.net/cover/8283/computational-genome-wide-identification-of-heat-shock-protein-genes-in-the-bovine-genome-version-1-peer-review-2-approved-1-approved-with-reservations-88283.webp)
Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]
F1000Research 2018, 7:1504 Last updated: 08 AUG 2021 RESEARCH ARTICLE Computational genome-wide identification of heat shock protein genes in the bovine genome [version 1; peer review: 2 approved, 1 approved with reservations] Oyeyemi O. Ajayi1,2, Sunday O. Peters3, Marcos De Donato2,4, Sunday O. Sowande5, Fidalis D.N. Mujibi6, Olanrewaju B. Morenikeji2,7, Bolaji N. Thomas 8, Matthew A. Adeleke 9, Ikhide G. Imumorin2,10,11 1Department of Animal Breeding and Genetics, Federal University of Agriculture, Abeokuta, Nigeria 2International Programs, College of Agriculture and Life Sciences, Cornell University, Ithaca, NY, 14853, USA 3Department of Animal Science, Berry College, Mount Berry, GA, 30149, USA 4Departamento Regional de Bioingenierias, Tecnologico de Monterrey, Escuela de Ingenieria y Ciencias, Queretaro, Mexico 5Department of Animal Production and Health, Federal University of Agriculture, Abeokuta, Nigeria 6Usomi Limited, Nairobi, Kenya 7Department of Animal Production and Health, Federal University of Technology, Akure, Nigeria 8Department of Biomedical Sciences, Rochester Institute of Technology, Rochester, NY, 14623, USA 9School of Life Sciences, University of KwaZulu-Natal, Durban, 4000, South Africa 10School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, 30032, USA 11African Institute of Bioscience Research and Training, Ibadan, Nigeria v1 First published: 20 Sep 2018, 7:1504 Open Peer Review https://doi.org/10.12688/f1000research.16058.1 Latest published: 20 Sep 2018, 7:1504 https://doi.org/10.12688/f1000research.16058.1 Reviewer Status Invited Reviewers Abstract Background: Heat shock proteins (HSPs) are molecular chaperones 1 2 3 known to bind and sequester client proteins under stress. Methods: To identify and better understand some of these proteins, version 1 we carried out a computational genome-wide survey of the bovine 20 Sep 2018 report report report genome. -

The B Vitamins Nicotinamide (B3) and Riboflavin (B2)
The B Vitamins Nicotinamide (B3) and Riboflavin (B2) Stimulate Metamorphosis in Larvae of the Deposit- Feeding Polychaete Capitella teleta: Implications for a Sensory Ligand-Gated Ion Channel Robert T. Burns1*, Jan A. Pechenik1, William J. Biggers2, Gia Scavo2, Christopher Lehman2 1 Department of Biology, Tufts University, Medford, Massachusetts, United States of America, 2 Department of Biology, Wilkes University, Wilkes-Barre, Pennsylvania, United States of America Abstract Marine sediments can contain B vitamins, presumably incorporated from settled, decaying phytoplankton and microorganisms associated with decomposition. Because B vitamins may be advantageous for the energetically intensive processes of metamorphosis, post-metamorphic growth, and reproduction, we tested several B vitamins to determine if they would stimulate larvae of the deposit-feeding polychaete Capitella teleta to settle and metamorphose. Nicotinamide and riboflavin individually stimulated larvae of C. teleta to settle and metamorphose, generally within 1–2 hours at nicotinamide concentrations as low as 3 mM and riboflavin concentrations as low as 50 mM. More than 80% of the larvae metamorphosed within 30 minutes at a nicotinamide concentration of 7 mM. The pyridine channel agonist pyrazinecarboxamide also stimulated metamorphosis at very low concentrations. In contrast, neither lumichrome, thiamine HCl, pyridoxine HCl, nor vitamin B12 stimulated larvae of C. teleta to metamorphose at concentrations as high as 500 mM. Larvae also did not metamorphose in response to either nicotinamide or pyrazinecarboxamide in calcium-free seawater or with the addition of 4-acetylpyridine, a competitive inhibitor of the pyridine receptor. Together, these results suggest that larvae of C. teleta are responding to nicotinamide and riboflavin via a chemosensory pyridine receptor similar to that previously reported to be present on crayfish chela and involved with food recognition. -

Neural Stem Cell-Derived Exosomes Revert HFD-Dependent Memory Impairment Via CREB-BDNF Signalling
International Journal of Molecular Sciences Article Neural Stem Cell-Derived Exosomes Revert HFD-Dependent Memory Impairment via CREB-BDNF Signalling Matteo Spinelli 1, Francesca Natale 1,2, Marco Rinaudo 1, Lucia Leone 1,2, Daniele Mezzogori 1, Salvatore Fusco 1,2,* and Claudio Grassi 1,2 1 Department of Neuroscience, Università Cattolica del Sacro Cuore, 00168 Rome, Italy; [email protected] (M.S.); [email protected] (F.N.); [email protected] (M.R.); [email protected] (L.L.); [email protected] (D.M.); [email protected] (C.G.) 2 Fondazione Policlinico Universitario Agostino Gemelli IRCCS, 00168 Rome, Italy * Correspondence: [email protected] Received: 6 October 2020; Accepted: 25 November 2020; Published: 26 November 2020 Abstract: Overnutrition and metabolic disorders impair cognitive functions through molecular mechanisms still poorly understood. In mice fed with a high fat diet (HFD) we analysed the expression of synaptic plasticity-related genes and the activation of cAMP response element-binding protein (CREB)-brain-derived neurotrophic factor (BDNF)-tropomyosin receptor kinase B (TrkB) signalling. We found that a HFD inhibited both CREB phosphorylation and the expression of a set of CREB target genes in the hippocampus. The intranasal administration of neural stem cell (NSC)-derived exosomes (exo-NSC) epigenetically restored the transcription of Bdnf, nNOS, Sirt1, Egr3, and RelA genes by inducing the recruitment of CREB on their regulatory sequences. Finally, exo-NSC administration rescued both BDNF signalling and memory in HFD mice. Collectively, our findings highlight novel mechanisms underlying HFD-related memory impairment and provide evidence of the potential therapeutic effect of exo-NSC against metabolic disease-related cognitive decline. -

Neurotransmitters-Drugs Andbrain Function.Pdf
Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Edited by R. A. Webster Department of Pharmacology, University College London, UK JOHN WILEY & SONS, LTD Chichester Á New York Á Weinheim Á Brisbane Á Singapore Á Toronto Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Copyright # 2001 by John Wiley & Sons Ltd. Bans Lane, Chichester, West Sussex PO19 1UD, UK National 01243 779777 International ++44) 1243 779777 e-mail +for orders and customer service enquiries): [email protected] Visit our Home Page on: http://www.wiley.co.uk or http://www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1P0LP,UK, without the permission in writing of the publisher. Other Wiley Editorial Oces John Wiley & Sons, Inc., 605 Third Avenue, New York, NY 10158-0012, USA WILEY-VCH Verlag GmbH, Pappelallee 3, D-69469 Weinheim, Germany John Wiley & Sons Australia, Ltd. -

Resistance to Ondansetron: Role of Pharmacogenetics in Post-Operative Nausea and Vomiting
The Egyptian Journal of Medical Human Genetics (2013) 14, 331–336 Ain Shams University The Egyptian Journal of Medical Human Genetics www.ejmhg.eg.net www.sciencedirect.com REVIEW Resistance to ondansetron: Role of pharmacogenetics in post-operative nausea and vomiting Kulsoom Farhat a,*, Muhammad Ismail b, Shabana Ali a, Anwar Kamal Pasha c a Army Medical College (National University of Sciences & Technology), Pakistan b Institute of Biomedical and Genetic Engineering, Pakistan c Combined Military Hospital, Rawalpindi, Pakistan Received 28 January 2013; accepted 17 March 2013 Available online 4 May 2013 KEYWORDS Abstract Post-operative nausea and vomiting is the most annoying and at the same time a danger- 5-Hydroxytryptamine type 3 ous side effect of general anaesthesia. Ondansetron is a routinely used anti emetic drug which is receptor antagonists; being administered by the trial and error principle. Though it did revolutionized the management Ondansetron; of this condition but by and large failed to completely eliminate the problem. Recently an important Post-operative nausea and factor possibly elucidating this failure is said to be the differing expression of genes controlling pro- vomiting; teins that are involved in transport and receptors related to this drug. Quite surprisingly these trans- Polymorphism; porter and receptor pathways have been found to be polymorphic and at the same time shown to be Pharmacogenetics related to efficacy of the drug. The differentiation between those responding to treatment and those not responding may pave a way to individualize treatment for emesis to a greater extent. This review highlights the pharmacogenetics related to this commonly used anti-emetic drug in anaesthesia. -

Investigation of Candidate Genes and Mechanisms Underlying Obesity
Prashanth et al. BMC Endocrine Disorders (2021) 21:80 https://doi.org/10.1186/s12902-021-00718-5 RESEARCH ARTICLE Open Access Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules G. Prashanth1 , Basavaraj Vastrad2 , Anandkumar Tengli3 , Chanabasayya Vastrad4* and Iranna Kotturshetti5 Abstract Background: Obesity associated type 2 diabetes mellitus is a metabolic disorder ; however, the etiology of obesity associated type 2 diabetes mellitus remains largely unknown. There is an urgent need to further broaden the understanding of the molecular mechanism associated in obesity associated type 2 diabetes mellitus. Methods: To screen the differentially expressed genes (DEGs) that might play essential roles in obesity associated type 2 diabetes mellitus, the publicly available expression profiling by high throughput sequencing data (GSE143319) was downloaded and screened for DEGs. Then, Gene Ontology (GO) and REACTOME pathway enrichment analysis were performed. The protein - protein interaction network, miRNA - target genes regulatory network and TF-target gene regulatory network were constructed and analyzed for identification of hub and target genes. The hub genes were validated by receiver operating characteristic (ROC) curve analysis and RT- PCR analysis. Finally, a molecular docking study was performed on over expressed proteins to predict the target small drug molecules. Results: A total of 820 DEGs were identified between -

Understanding Allergic Multimorbidity Within the Non-Eosinophilic
University of Groningen Understanding allergic multimorbidity within the non-eosinophilic interactome Aguilar, Daniel; Lemonnier, Nathanael; Koppelman, Gerard H; Melén, Erik; Oliva, Baldo; Pinart, Mariona; Guerra, Stefano; Bousquet, Jean; Anto, Josep M Published in: PLoS ONE DOI: 10.1371/journal.pone.0224448 IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2019 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Aguilar, D., Lemonnier, N., Koppelman, G. H., Melén, E., Oliva, B., Pinart, M., Guerra, S., Bousquet, J., & Anto, J. M. (2019). Understanding allergic multimorbidity within the non-eosinophilic interactome. PLoS ONE, 14(11), [e0224448]. https://doi.org/10.1371/journal.pone.0224448 Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 26-12-2020 RESEARCH ARTICLE Understanding allergic multimorbidity within the non-eosinophilic interactome 1,2,3 4 5,6 7 Daniel AguilarID *, Nathanael LemonnierID , Gerard H. -

Supplementary Table 1. Pain and PTSS Associated Genes (N = 604
Supplementary Table 1. Pain and PTSS associated genes (n = 604) compiled from three established pain gene databases (PainNetworks,[61] Algynomics,[52] and PainGenes[42]) and one PTSS gene database (PTSDgene[88]). These genes were used in in silico analyses aimed at identifying miRNA that are predicted to preferentially target this list genes vs. a random set of genes (of the same length). ABCC4 ACE2 ACHE ACPP ACSL1 ADAM11 ADAMTS5 ADCY5 ADCYAP1 ADCYAP1R1 ADM ADORA2A ADORA2B ADRA1A ADRA1B ADRA1D ADRA2A ADRA2C ADRB1 ADRB2 ADRB3 ADRBK1 ADRBK2 AGTR2 ALOX12 ANO1 ANO3 APOE APP AQP1 AQP4 ARL5B ARRB1 ARRB2 ASIC1 ASIC2 ATF1 ATF3 ATF6B ATP1A1 ATP1B3 ATP2B1 ATP6V1A ATP6V1B2 ATP6V1G2 AVPR1A AVPR2 BACE1 BAMBI BDKRB2 BDNF BHLHE22 BTG2 CA8 CACNA1A CACNA1B CACNA1C CACNA1E CACNA1G CACNA1H CACNA2D1 CACNA2D2 CACNA2D3 CACNB3 CACNG2 CALB1 CALCRL CALM2 CAMK2A CAMK2B CAMK4 CAT CCK CCKAR CCKBR CCL2 CCL3 CCL4 CCR1 CCR7 CD274 CD38 CD4 CD40 CDH11 CDK5 CDK5R1 CDKN1A CHRM1 CHRM2 CHRM3 CHRM5 CHRNA5 CHRNA7 CHRNB2 CHRNB4 CHUK CLCN6 CLOCK CNGA3 CNR1 COL11A2 COL9A1 COMT COQ10A CPN1 CPS1 CREB1 CRH CRHBP CRHR1 CRHR2 CRIP2 CRYAA CSF2 CSF2RB CSK CSMD1 CSNK1A1 CSNK1E CTSB CTSS CX3CL1 CXCL5 CXCR3 CXCR4 CYBB CYP19A1 CYP2D6 CYP3A4 DAB1 DAO DBH DBI DICER1 DISC1 DLG2 DLG4 DPCR1 DPP4 DRD1 DRD2 DRD3 DRD4 DRGX DTNBP1 DUSP6 ECE2 EDN1 EDNRA EDNRB EFNB1 EFNB2 EGF EGFR EGR1 EGR3 ENPP2 EPB41L2 EPHB1 EPHB2 EPHB3 EPHB4 EPHB6 EPHX2 ERBB2 ERBB4 EREG ESR1 ESR2 ETV1 EZR F2R F2RL1 F2RL2 FAAH FAM19A4 FGF2 FKBP5 FLOT1 FMR1 FOS FOSB FOSL2 FOXN1 FRMPD4 FSTL1 FYN GABARAPL1 GABBR1 GABBR2 GABRA2 GABRA4 -
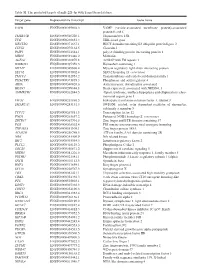
Table SI. the Predicted Targets of Mir-221-3P with Targetscan Database
Table SI. The predicted targets of miR-221-3p with TargetScan database. Target gene Representative transcript Gene name VAPB ENST00000395802.3 VAMP (vesicle-associated membrane protein)-associated protein B and C TMSB15B ENST00000540220.1 Thymosin beta 15B TFG ENST00000240851.4 TRK-fused gene HECTD2 ENST00000371667.1 HECT domain containing E3 ubiquitin protein ligase 2 CLVS2 ENST00000275162.5 Clavesin 2 PAIP1 ENST00000514514.1 poly(A) binding protein interacting protein 1 MIDN ENST00000591446.2 Midnolin AGFG1 ENST00000310078.8 ArfGAP with FG repeats 1 HMBOX1 ENST00000397358.3 Homeobox containing 1 MYLIP ENST00000349606.4 Myosin regulatory light chain interacting protein SEC62 ENST00000337002.4 SEC62 homolog (S. cerevisiae) TMCC1 ENST00000432054.2 Transmembrane and coiled-coil domain family 1 PHACTR4 ENST00000373839.3 Phosphatase and actin regulator 4 AIDA ENST00000340020.6 Axin interactor, dorsalization associated BEAN1 ENST00000299694.8 Brain expressed, associated with NEDD4, 1 AMMECR1 ENST00000262844.5 Alport syndrome, midface hypoplasia and elliptocytosis chro- mosomal region gene 1 EIF3J ENST00000261868.5 Eukaryotic translation initiation factor 3, subunit J SMARCA5 ENST00000283131.3 SWI/SNF related, actin dependent regulator of chromatin, subfamily a, member 5 TCF12 ENST00000267811.5 Transcription factor 12 PNO1 ENST00000263657.2 Partner of NOB1 homolog (S. cerevisiae) ZBTB37 ENST00000367701.5 Zinc finger and BTB domain containing 37 FOS ENST00000303562.4 FBJ murine osteosarcoma viral oncogene homolog ZNF385A ENST00000551109.1 Zinc -

The 5-HT3AB Receptor Shows an A3B2 Stoichiometry at the Plasma Membrane
Biophysical Journal Volume 105 August 2013 887–898 887 The 5-HT3AB Receptor Shows an A3B2 Stoichiometry at the Plasma Membrane Timothy F. Miles,† Dennis A. Dougherty,‡ and Henry A. Lester†* †Division of Biology and Biological Engineering and ‡Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California ABSTRACT The 5-HT3AB receptor is the best-characterized heteropentameric 5-HT3 receptor. Under conditions of heterol- ogous expression, the 5-HT3AB receptor shows a single functionally resolvable population, suggesting the presence of a unique subunit stoichiometry; however, conflicting previous reports have suggested two different possible stoichiometries. Here we isolate plasma membrane sheets containing assembled receptors from individual HEK293T cells. We then determine the stoi- chiometry of 5-HT3AB receptors on the plasma membrane by fluorescence methods, employing meCFP- and meYFP-labeled A and B subunits. Over a wide range of cDNA transfection ratios, fluorescence intensity ratios are closest to values that corre- spond to a subunit ratio of A3B2.Fo¨rster resonance energy transfer (family FRET) efficiencies provide minor corrections (3–6%) to the subunit ratios and provide independent support for a predominantly A3B2 stoichiometry on the plasma membrane sheets. Twin FRET efficiencies support these data, also suggesting that the two B subunits are nonadjacent in most of the het- eropentamers. The high-frequency variant HTR3B p.Y129S (c.386A>C, rs11767445), linked to psychiatric disease, also forms A3B2 receptors on the plasma membrane. The 5-HT3B Y129S, subunit incorporates in a slightly (11–14%) more efficient manner than the common variant. In general, most of the subunits reside within the cell. -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. The CX3CR1/CX3CL1 Axis Drives the Migration and Maturation of Oligodendroglia in the Central Nervous System Catriona Ford Thesis Submitted for the Degree of Doctor of Philosophy The University of Edinburgh 2017 Abstract In the central nervous system, the axons of neurons are protected from damage and aided in electrical conductivity by the myelin sheath, a complex of proteins and lipids formed by oligodendrocytes. Loss or damage to the myelin sheath may result in impairment of electrical axonal conduction and eventually to neuronal death. Such demyelination is responsible, at least in part, for the disabling neurodegeneration observed in pathologies such as Multiple Sclerosis (MS) and Spinal Cord Injury. In the regenerative process of remyelination, oligodendrocyte precursor cells (OPCs), the resident glial stem cell population of the adult CNS, migrate toward the injury site, proliferate and differentiate into adult oligodendrocytes which subsequently reform the myelin sheath.