Delineating the Underlying Molecular Mechanisms and Key Genes Involved in Metastasis of Colorectal Cancer Via Bioinformatics Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phosphorylation of Synaptojanin Differentially Regulates Endocytosis of Functionally Distinct Synaptic Vesicle Pools
8882 • The Journal of Neuroscience, August 24, 2016 • 36(34):8882–8894 Cellular/Molecular Phosphorylation of Synaptojanin Differentially Regulates Endocytosis of Functionally Distinct Synaptic Vesicle Pools X Junhua Geng,1* Liping Wang,1,2* Joo Yeun Lee,1,4 XChun-Kan Chen,1 and Karen T. Chang1,3,4 1Zilkha Neurogenetic Institute, 2Department of Biochemistry and Molecular Biology, and 3Department of Cell and Neurobiology, Keck School of Medicine, University of Southern California, Los Angeles, California 90089, and 4Neuroscience Graduate Program, University of Southern California, Los Angeles, California 90089 The rapid replenishment of synaptic vesicles through endocytosis is crucial for sustaining synaptic transmission during intense neuronal activity. Synaptojanin (Synj), a phosphoinositide phosphatase, is known to play an important role in vesicle recycling by promoting the uncoating of clathrin following synaptic vesicle uptake. Synj has been shown to be a substrate of the minibrain (Mnb) kinase, a fly homolog of the dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A); however, the functional impacts of Synj phosphorylation by Mnb are not well understood. Here we identify that Mnb phosphorylates Synj at S1029 in Drosophila. We find that phosphorylation of Synj at S1029 enhances Synj phosphatase activity, alters interaction between Synj and endophilin, and promotes efficient endocytosis of the active cycling vesicle pool (also referred to as exo-endo cycling pool) at the expense of reserve pool vesicle endocytosis. Dephosphorylated Synj, on the other hand, is deficient in the endocytosis of the active recycling pool vesicles but maintains reserve pool vesicle endocytosis to restore total vesicle pool size and sustain synaptic transmission. Together, our findings reveal a novel role for Synj in modulating reserve pool vesicle endocytosis and further indicate that dynamic phosphorylation and dephosphorylation of Synj differentially maintain endocytosis of distinct functional synaptic vesicle pools. -

Intersectin 1 Forms Complexes with SGIP1 and Reps1 in Clathrin-Coated
Biochemical and Biophysical Research Communications 402 (2010) 408–413 Contents lists available at ScienceDirect Biochemical and Biophysical Research Communications journal homepage: www.elsevier.com/locate/ybbrc Intersectin 1 forms complexes with SGIP1 and Reps1 in clathrin-coated pits ⇑ Oleksandr Dergai a, ,1, Olga Novokhatska a,1, Mykola Dergai a, Inessa Skrypkina a, Liudmyla Tsyba a, Jacques Moreau b, Alla Rynditch a a Department of Functional Genomics, Institute of Molecular Biology and Genetics, NASU, 150 Zabolotnogo Street, 03680 Kyiv, Ukraine b Molecular Mechanisms of Development, Jacques Monod Institute, Development and Neurobiology Program, UMR7592 CNRS – Paris Diderot University, 15 rue Hélène Brion, 75205 Paris Cedex 13, France article info abstract Article history: Intersectin 1 (ITSN1) is an evolutionarily conserved adaptor protein involved in clathrin-mediated endo- Received 7 October 2010 cytosis, cellular signaling and cytoskeleton rearrangement. ITSN1 gene is located on human chromosome Available online 12 October 2010 21 in Down syndrome critical region. Several studies confirmed role of ITSN1 in Down syndrome pheno- type. Here we report the identification of novel interconnections in the interaction network of this endo- Keywords: cytic adaptor. We show that the membrane-deforming protein SGIP1 (Src homology 3-domain growth Endocytosis factor receptor-bound 2-like (endophilin) interacting protein 1) and the signaling adaptor Reps1 (RalBP Adaptor proteins associated Eps15-homology domain protein) interact with ITSN1 in vivo. Both interactions are mediated Intersectin 1 by the SH3 domains of ITSN1 and proline-rich motifs of protein partners. Moreover complexes compris- Protein interactions SGIP1 ing SGIP1, Reps1 and ITSN1 have been identified. We also identified new interactions between SGIP1, Reps1 Reps1 and the BAR (Bin/amphiphysin/Rvs) domain-containing protein amphiphysin 1. -

Supplemental Figure 1. Vimentin
Double mutant specific genes Transcript gene_assignment Gene Symbol RefSeq FDR Fold- FDR Fold- FDR Fold- ID (single vs. Change (double Change (double Change wt) (single vs. wt) (double vs. single) (double vs. wt) vs. wt) vs. single) 10485013 BC085239 // 1110051M20Rik // RIKEN cDNA 1110051M20 gene // 2 E1 // 228356 /// NM 1110051M20Ri BC085239 0.164013 -1.38517 0.0345128 -2.24228 0.154535 -1.61877 k 10358717 NM_197990 // 1700025G04Rik // RIKEN cDNA 1700025G04 gene // 1 G2 // 69399 /// BC 1700025G04Rik NM_197990 0.142593 -1.37878 0.0212926 -3.13385 0.093068 -2.27291 10358713 NM_197990 // 1700025G04Rik // RIKEN cDNA 1700025G04 gene // 1 G2 // 69399 1700025G04Rik NM_197990 0.0655213 -1.71563 0.0222468 -2.32498 0.166843 -1.35517 10481312 NM_027283 // 1700026L06Rik // RIKEN cDNA 1700026L06 gene // 2 A3 // 69987 /// EN 1700026L06Rik NM_027283 0.0503754 -1.46385 0.0140999 -2.19537 0.0825609 -1.49972 10351465 BC150846 // 1700084C01Rik // RIKEN cDNA 1700084C01 gene // 1 H3 // 78465 /// NM_ 1700084C01Rik BC150846 0.107391 -1.5916 0.0385418 -2.05801 0.295457 -1.29305 10569654 AK007416 // 1810010D01Rik // RIKEN cDNA 1810010D01 gene // 7 F5 // 381935 /// XR 1810010D01Rik AK007416 0.145576 1.69432 0.0476957 2.51662 0.288571 1.48533 10508883 NM_001083916 // 1810019J16Rik // RIKEN cDNA 1810019J16 gene // 4 D2.3 // 69073 / 1810019J16Rik NM_001083916 0.0533206 1.57139 0.0145433 2.56417 0.0836674 1.63179 10585282 ENSMUST00000050829 // 2010007H06Rik // RIKEN cDNA 2010007H06 gene // --- // 6984 2010007H06Rik ENSMUST00000050829 0.129914 -1.71998 0.0434862 -2.51672 -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Use of Genomic Tools to Discover the Cause of Champagne Dilution Coat Color in Horses and to Map the Genetic Cause of Extreme Lordosis in American Saddlebred Horses
University of Kentucky UKnowledge Theses and Dissertations--Veterinary Science Veterinary Science 2014 USE OF GENOMIC TOOLS TO DISCOVER THE CAUSE OF CHAMPAGNE DILUTION COAT COLOR IN HORSES AND TO MAP THE GENETIC CAUSE OF EXTREME LORDOSIS IN AMERICAN SADDLEBRED HORSES Deborah G. Cook University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Cook, Deborah G., "USE OF GENOMIC TOOLS TO DISCOVER THE CAUSE OF CHAMPAGNE DILUTION COAT COLOR IN HORSES AND TO MAP THE GENETIC CAUSE OF EXTREME LORDOSIS IN AMERICAN SADDLEBRED HORSES" (2014). Theses and Dissertations--Veterinary Science. 15. https://uknowledge.uky.edu/gluck_etds/15 This Doctoral Dissertation is brought to you for free and open access by the Veterinary Science at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Veterinary Science by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -
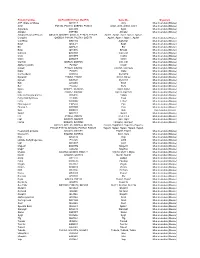
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Mutations in Pi(3,5)P2 Signaling and Neurodegeneration in Mouse and Human
MUTATIONS IN PI(3,5)P2 SIGNALING AND NEURODEGENERATION IN MOUSE AND HUMAN by Clement Y. Chow A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Human Genetics) in The University of Michigan 2008 Doctoral Committee: Professor Miriam H. Meisler, Chair Professor Sally A. Camper Professor David Ginsburg Associate Professor David C. Kohrman Assistant Professor Geoffrey G. Murphy Clement Y. Chow 2008 To my wife, Candace For all her love and support ii ACKNOWLEDGEMENTS I would first like to thank my mentor, Miriam Meisler. She is the consummate scientist. Miriam is inquisitive and interested in a wide range of topics. This trait has taught me to always ask the right questions and be skeptical of conclusions that are not supported by data. Miriam’s unending desire to make each oral presentation perfect drove me to improve my own skills. I came to this lab without any public presentation skills, but I am now greatly improved in my presentation abilities, a skill crucial for a successful scientific career. Miriam is a perfectionist when it comes to writing and always encouraged me to do the same. I will continue to strive to perfect my writing abilities. I came to the lab wanting to positionally clone a mouse mutant. I told Miriam that I would stay in the lab if she allowed me to do so. She was supportive and excited from the beginning, allowing me to pursue a project with an unknown future. This allowed me to learn, first hand, many aspects of genetics that such a project provides. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Comparative Analysis of Human Chromosome 7Q21 and Mouse
Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Comparative analysis of human chromosome 7q21 and mouse proximal chromosome 6 reveals a placental-specific imprinted gene, TFPI2/Tfpi2, which requires EHMT2 and EED for allelic-silencing David Monk,1,6 Alexandre Wagschal,2 Philippe Arnaud,2 Pari-Sima Mu¨ller,3 Layla Parker-Katiraee,4 Déborah Bourc’his,5 Stephen W. Scherer,4 Robert Feil,2 Philip Stanier,1 and Gudrun E. Moore1 1Institute of Child Health, London WC1N 1EH, United Kingdom; 2Institute of Molecular Genetics, CNRS UMR-5535 and University of Montpellier-II, 34293 Montpellier, France; 3Sir William Dunn School of Pathology, University of Oxford, Oxford OX1 3RE, United Kingdom; 4Center for Applied Genomics, The Hospital for Sick Children, Toronto M5G 1L7, Canada; 5Inserm U741, F-75251 Paris Cedex 05, France Genomic imprinting is a developmentally important mechanism that involves both differential DNA methylation and allelic histone modifications. Through detailed comparative characterization, a large imprinted domain mapping to chromosome 7q21 in humans and proximal chromosome 6 in mice was redefined. This domain is organized around a maternally methylated CpG island comprising the promoters of the adjacent PEG10 and SGCE imprinted genes. Examination of Dnmt3l−/+ conceptuses shows that imprinted expression for all genes of the cluster depends upon the germline methylation at this putative “imprinting control region” (ICR). Similarly as for other ICRs, we find its DNA-methylated allele to be associated with trimethylation of lysine 9 on histone H3 (H3K9me3) and trimethylation of lysine 20 on histone H4 (H4K20me3), whereas the transcriptionally active paternal allele is enriched in H3K4me2 and H3K9 acetylation.