United States Patent Office Patented Oct
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Stock Solutions
STOCK SOLUTIONS 5 M NaCl (F.W. 58.44) 500 ml: 146.1 g NaCl ~350 ml H2O Dissolve, then bring up to volume with H2O Sterilize by autoclaving (15 minutes) 1 M Tris, pH 8 (F.W. 121.1) 500 ml: 60.55 g Trizma base ~400 ml H2O Approximately 21.1 ml concentrated HCl (Use pH meter) Bring up to volume with H2O Sterilize by autoclaving (15 minutes) 1 M Tris, pH 7.6 (F.W. 121.1) 500 ml: 60.55 g Trizma base ~400 ml H2O Approximately 28.5 ml concentrated HCl (Use pH meter) Bring up to volume with H2O Sterilize by autoclaving (15 minutes) 1 M MgCl2 (F.W. 203.30) 100 ml: 20.33 g MgCl2 ~70 ml H2O Dissolve, then bring up to volume with H2O Sterilize by autoclaving (15 minutes) 1 M CaCl2 (F.W. 147.02) 100 ml: 14.70 g CaCl2 ~90 ml H2O Dissolve, then bring up to volume with H2O Sterilization not required 1 M MgSO4 (F.W. 246.47) 100 ml: 24.65 g MgSO4 ~75 ml H2O Dissolve, then bring up to volume with H2O Sterilize by autoclaving (15 minutes) 0.1 M MgCl2 (F.W. 203.30) 100 ml: 2.03 g MgCl2 ~95 ml H2O Dissolve, then bring up to volume with H2O Sterilize by autoclaving (15 minutes) 3 M NaOH (F.W. 40.00) 100 ml: 12.0 g NaOH ~80 ml H2O Dissolve, then bring up to volume with H2O Sterilization not required 2 M Sorbitol (F.W. 182.2) 500 ml: 182.2 g Sorbitol ~300 ml H2O Dissolve, then bring up to volume with H2O Sterilization by filtration 5 M Potassium acetate (F.W. -

NON-HAZARDOUS CHEMICALS May Be Disposed of Via Sanitary Sewer Or Solid Waste
NON-HAZARDOUS CHEMICALS May Be Disposed Of Via Sanitary Sewer or Solid Waste (+)-A-TOCOPHEROL ACID SUCCINATE (+,-)-VERAPAMIL, HYDROCHLORIDE 1-AMINOANTHRAQUINONE 1-AMINO-1-CYCLOHEXANECARBOXYLIC ACID 1-BROMOOCTADECANE 1-CARBOXYNAPHTHALENE 1-DECENE 1-HYDROXYANTHRAQUINONE 1-METHYL-4-PHENYL-1,2,5,6-TETRAHYDROPYRIDINE HYDROCHLORIDE 1-NONENE 1-TETRADECENE 1-THIO-B-D-GLUCOSE 1-TRIDECENE 1-UNDECENE 2-ACETAMIDO-1-AZIDO-1,2-DIDEOXY-B-D-GLYCOPYRANOSE 2-ACETAMIDOACRYLIC ACID 2-AMINO-4-CHLOROBENZOTHIAZOLE 2-AMINO-2-(HYDROXY METHYL)-1,3-PROPONEDIOL 2-AMINOBENZOTHIAZOLE 2-AMINOIMIDAZOLE 2-AMINO-5-METHYLBENZENESULFONIC ACID 2-AMINOPURINE 2-ANILINOETHANOL 2-BUTENE-1,4-DIOL 2-CHLOROBENZYLALCOHOL 2-DEOXYCYTIDINE 5-MONOPHOSPHATE 2-DEOXY-D-GLUCOSE 2-DEOXY-D-RIBOSE 2'-DEOXYURIDINE 2'-DEOXYURIDINE 5'-MONOPHOSPHATE 2-HYDROETHYL ACETATE 2-HYDROXY-4-(METHYLTHIO)BUTYRIC ACID 2-METHYLFLUORENE 2-METHYL-2-THIOPSEUDOUREA SULFATE 2-MORPHOLINOETHANESULFONIC ACID 2-NAPHTHOIC ACID 2-OXYGLUTARIC ACID 2-PHENYLPROPIONIC ACID 2-PYRIDINEALDOXIME METHIODIDE 2-STEP CHEMISTRY STEP 1 PART D 2-STEP CHEMISTRY STEP 2 PART A 2-THIOLHISTIDINE 2-THIOPHENECARBOXYLIC ACID 2-THIOPHENECARBOXYLIC HYDRAZIDE 3-ACETYLINDOLE 3-AMINO-1,2,4-TRIAZINE 3-AMINO-L-TYROSINE DIHYDROCHLORIDE MONOHYDRATE 3-CARBETHOXY-2-PIPERIDONE 3-CHLOROCYCLOBUTANONE SOLUTION 3-CHLORO-2-NITROBENZOIC ACID 3-(DIETHYLAMINO)-7-[[P-(DIMETHYLAMINO)PHENYL]AZO]-5-PHENAZINIUM CHLORIDE 3-HYDROXYTROSINE 1 9/26/2005 NON-HAZARDOUS CHEMICALS May Be Disposed Of Via Sanitary Sewer or Solid Waste 3-HYDROXYTYRAMINE HYDROCHLORIDE 3-METHYL-1-PHENYL-2-PYRAZOLIN-5-ONE -

Potassium Acetate
OKI GROUNDWATER COMMITTEE June 3, 2009 - 10:00 A.M. OKI Board Room 720 East Pete Rose Way (at the corner of Eggleston Avenue)* AGENDA 1. Welcome/Introductions 2. Announcements 3. OKI Staff Updates 4. Update on Local Groundwater Management Efforts Frank Bell, Mike Heuer, Greg Stanley, Bruce Whitteberry, Tom Yeager 5. The St. Clair Township Comprehensive Plan: Protecting Source Water and Community Character Jane Wittke, OKI 6. The Groundwater Implications of Geothermal Systems: Geothermal Systems in Ohio Russell Smith, Ohio Department of Health and Geothermal System Construction Jacob Crabtree, Crabtree Drilling Company 7. Other Business ADJOURNMENT ** SEE the MAP and DIRECTIONS on the REVERSE HARD COPY (or in separate attachment to email) Mill Creek Headwaters Project Funding Federal Funds: $498,000 from Ohio EPA under Section 319 of the Clean Water Act Local Match: $472,600 from West Chester Township’s conservation easements and project partners’ in-kind services Mill Creek Headwaters Project Purposes 1. Educate public on how to reduce water pollution 2. Evaluate best management practices 3. Reduce nonpoint source pollution by installing: • 400 feet of stabilized streambank • 3,000 feet of restored riparian corridor • 18 acres of restored floodplain wetland Project Partners •West Chester Township • OKI Regional Council of Governments • Butler Soil and Water Conservation District • Mill Creek Watershed Council of Communities • Butler County Engineer’s Office • Butler County Water and Sewer Department • Greenacres Foundation • University -

UNITED STATES PATENT Office
Patented Mar. 7, 1950 2499,924 UNITED STATES PATENT office 2,499,924 HIGH VIscosiTY PoLYVINYL Alcohol Edward Lavin, Springfield, Mass, assigner to Shawinigan Resins Corporation, Springfield, Mass, a corporation of Massachusetts No Drawing. Appliention November 20, 1946, Serial No. 711,207 4 Claims. (CL 260-91s), 2 This invention relates to a process for pre The process employed in preparing the prod paring polyvinyl alcohols having high viscosities. lucts set forth in the examples in Tables A and B Hydrolyzed polyvinyl acetates are useful for comprises dissolving the polyvinyl acetate in the many purposes. However, for some uses e. g., toluene and then adding the methanol containing as thickening agents, protective colloids and the the sulfuric acid (98% HaSO4) dissolved therein. like, it is desirable that such materials possess The charge is prepared in a suitable apparatus materially higher viscosities. provided with an agitator and a water-cooled It is an object of this invention to provide a return condenser. The hydrolysis is carried out process for preparing partially hydrolyzed poly by heating the charge at its reflux temperature vinyl acetates having an unusually high viscosity. O and continuing the heating under these condi A particular object of this invention is to pro tions until the desired degree of hydrolysis is vide a process for preparing high viscosity poly effected. The reaction mixture is then cooled vinyl alcohols having an acetate content of 15-40 and the hydrolyzed product which is in the form per cent by weight, calculated as polyvinyl of...a precipitate is allowed to settle. Thereafter acetate. -
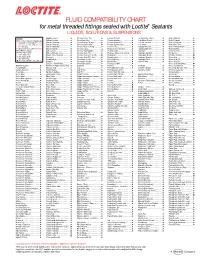
FLUID COMPATIBILITY CHART for Metal Threaded Fittings Sealed with Loctite¨ Sealants LIQUIDS, SOLUTIONS & SUSPENSIONS
FLUID COMPATIBILITY CHART for metal threaded fittings sealed with Loctite® Sealants LIQUIDS, SOLUTIONS & SUSPENSIONS LEGEND: Bagasse Fibers.......................... Chlorobenzene Dry ................... Ferrous Chloride ...................... Ion Exclusion Glycol ................. Nickel Chloride.......................... All Loctite® Anaerobic Sealants are Barium Acetate ........................ Chloroform Dry......................... Ferrous Oxalate......................... Irish Moss Slurry...................... Nickel Cyanide ......................... Compatible Including #242®, 243, Barium Carbonate..................... Chloroformate Methyl............... Ferrous Sulfate10%.................. Iron Ore Taconite ..................... Nickel Fluoborate ..................... 542, 545, 565, 567, 569, 571, 572, Barium Chloride........................ Chlorosulfonic Acid .................. Ferrous Sulfate (Sat)................. Iron Oxide ................................ Nickel Ore Fines ....................... 577, 580, 592 Barium Hydroxide..................... Chrome Acid Cleaning .............. Fertilizer Sol ............................. Isobutyl Alcohol ....................... Nickel Plating Bright ................. † Use Loctite® #270, 271™, 277, 554 Barium Sulfate.......................... Chrome Liquor.......................... Flotation Concentrates.............. Isobutyraldehyde ..................... Nickel Sulfate ........................... Not Recommended Battery Acid .............................. Chrome Plating -

Production of Low-Cost Acetate Deicers from Biomass and Industrial Wastes
Production of Low-Cost Acetate Deicers from Biomass and Industrial Wastes Shang-Tian Yang and Zuwei Jin, The Ohio State University Brian H. ChoUar, Federal Highway Administration Calcium magnesium acetate (CMA), a mixture of calcium 1 rom 10 million to 14 million tons of road salt are acetate and magnesium acetate, is used as an environmen• I used annually in the United States and Canada. tally benign roadway deicer. The present commercial F Salt is an extremely effective snow and ice con• CMA deicer made from glacial acetic acid and dolomitic trol agent and is relatively inexpensive. However, a lime or limestone is more expensive than salt and other study in New York State showed that although 1 ton of deicers. Also, a liquid potassium acetate deicer is used to road salt cost only $25, it caused more than $1,400 in replace urea and glycol in airport runway deicing. Two al• damage (1). Salt is corrosive to concrete and metals ternative low-cost methods to produce these acetate de- used in the nation's infrastructure, is harmful to road• icers from cheap feedstocks, such as biomass and side vegetation, and poses serious threats to environ• industrial wastes, were studied. CMA deicers produced ment and ground-water quality in some regions (2). from cheese whey by fermentation and extraction were FHWA spends about $12.5 billion annually, a sub• tested for their acetate content and deicing property. The stantial portion of which is used to rebuild and resur• CMA solid sample obtained from extraction of the acetic face highways and bridges damaged by salt corrosion. -

United States Patent Office Patented Sept
3,274,105 United States Patent Office Patented Sept. 20, 1966 2 Such as carbon disulfide and percaptains). Nevertheless, 3,274,05 Within the above-mentioned limits, it is advisable to FIRE EXTENGUISHENG COMPOSITION choose the concentration of the solution according to the Norbert Mevel, Antony, France, assignor to Societe d’Etudes Chimiques poeir distrie et l'Agriculare nature of the fires to be dealt with: for fires of high boil S.E.C.P.I.A., Paris, France ing hydrocarbons and those of a large number of chem No Drawing. Filed July 5, 1963, Ser. No. 295,213 icals Such as alcohol, aldehydes, amines, petroleum ether, Claims priority, application France, Aag. 3, 1962, ethyl ether, etc., solutions containing from 300 to 400 905,979, Patent 1,338,454 g./l. of potassium acetate give the best results. But for 10 Clains. (C. 252-2) fires of low boiling hydrocarbons such as gasoline, solu O tions containing from about 500 to 600 g./l. are more This invention relates to fire extinguishing Solutions for effective to obtain complete extinction. use against dry fires and fires particularly difficult to ex It has also been found that by adding tetrapotassium tinguish such as those of hydrocarbons or inflammable pyrophosphate to acqueous solutions of potassium acetate, chemicals and mixed fires, that is to say, fires of con the extinguishing effect is considerably increased and that bustible solid substances (wood, paper, rags, etc.) soaked 5 the same results are obtained with a lower acetate con for example with hydrocarbons or chemicals. Centration. For example, a solution containing 300 g./I. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Chemical Compatibility Storage Group
CHEMICAL SEGREGATION Chemicals are to be segregated into 11 different categories depending on the compatibility of that chemical with other chemicals The Storage Groups are as follows: Group A – Compatible Organic Acids Group B – Compatible Pyrophoric & Water Reactive Materials Group C – Compatible Inorganic Bases Group D – Compatible Organic Acids Group E – Compatible Oxidizers including Peroxides Group F– Compatible Inorganic Acids not including Oxidizers or Combustible Group G – Not Intrinsically Reactive or Flammable or Combustible Group J* – Poison Compressed Gases Group K* – Compatible Explosive or other highly Unstable Material Group L – Non-Reactive Flammable and Combustible, including solvents Group X* – Incompatible with ALL other storage groups The following is a list of chemicals and their compatibility storage codes. This is not a complete list of chemicals, but is provided to give examples of each storage group: Storage Group A 94‐75‐7 2,4‐D (2,4‐Dichlorophenoxyacetic acid) 94‐82‐6 2,4‐DB 609-99-4 3,5-Dinitrosalicylic acid 64‐19‐7 Acetic acid (Flammable liquid @ 102°F avoid alcohols, Amines, ox agents see SDS) 631-61-8 Acetic acid, Ammonium salt (Ammonium acetate) 108-24-7 Acetic anhydride (Flammable liquid @102°F avoid alcohols see SDS) 79‐10‐7 Acrylic acid Peroxide Former 65‐85‐0 Benzoic acid 98‐07‐7 Benzotrichloride 98‐88‐4 Benzoyl chloride 107-92-6 Butyric Acid 115‐28‐6 Chlorendic acid 79‐11‐8 Chloroacetic acid 627‐11‐2 Chloroethyl chloroformate 77‐92‐9 Citric acid 5949-29-1 Citric acid monohydrate 57-00-1 Creatine 20624-25-3 -

Topic: Environmental Field Evaluation of Potassium Acetate (TRIG NS 526)
Topic: Environmental Field Evaluation of Potassium Acetate (TRIG NS 526) Date: June 29, 2018 Prepared for: Beth Klemann Prepared by: Marilee Tuite, x3797 Resources searched: MnDOT Library catalog, TRID, RiP, ASCE database, PubMed Summary: The results below are relevant to potassium acetate, but the studies review other deicers and aren’t exclusive to potassium acetate. A few results specific to airport runoff are listed at the end. There seems to be more literature about calcium magnesium acetate’s environmental impacts than potassium acetate. Full-text links are provided where possible. Please contact the library if you would like to request full text through inter-library loan (ILL). Results Title: Environmental Risks of Snow and Ice Control Materials Source: chapter 10 (pp. 180-210) by Laura Fay et al in book Sustainable Winter Road Operations (April 2018) Abstract: As abrasives and deicer products are widely used for snow and ice control, the past decade has seen increased concerns over the detrimental impacts on the surrounding environment. Generally, these snow and ice control materials can cause impacts to air quality, water quality, soil, flora, fauna, and human health. The added cost of clean‐up and the paradigm shift from reactive to proactive snow and ice control strategies have resulted in growing use of chemicals instead of abrasives. Therefore, there is a need to better understand and assess the environmental impacts of deicers, in an effort to conduct sustainable winter road maintenance operations in an environmentally and fiscally responsible manner. This chapter will discuss relevant background information and provide a review of the environmental impacts of snow and ice control materials, including abrasives, chlorides, acetates and formates, glycols, urea, and agro‐based deicers, according to a survey of published work particularly those over the last two decades. -

Fire Diamond HFRS Ratings
Fire Diamond HFRS Ratings CHEMICAL H F R Special Hazards 1-aminobenzotriazole 0 1 0 1-butanol 2 3 0 1-heptanesulfonic acid sodium salt not hazardous 1-methylimidazole 3 2 1 corrosive, toxic 1-methyl-2pyrrolidinone, anhydrous 2 2 1 1-propanol 2 3 0 1-pyrrolidinecarbonyl chloride 3 1 0 1,1,1,3,3,3-Hexamethyldisilazane 98% 332corrosive 1,2-dibromo-3-chloropropane regulated carcinogen 1,2-dichloroethane 1 3 1 1,2-dimethoxyethane 1 3 0 peroxide former 1,2-propanediol 0 1 0 1,2,3-heptanetriol 2 0 0 1,2,4-trichlorobenzene 2 1 0 1,3-butadiene regulated carcinogen 1,4-diazabicyclo(2,2,2)octane 2 2 1 flammable 1,4-dioxane 2 3 1 carcinogen, may form explosive mixture in air 1,6-hexanediol 1 0 0 1,10-phenanthroline 2 1 0 2-acetylaminofluorene regulated carcinogen, skin absorbtion 2-aminobenzamide 1 1 1 2-aminoethanol 3 2 0 corrosive 2-Aminoethylisothiouronium bromide hydrobromide 2 0 0 2-aminopurine 1 0 0 2-amino-2-methyl-1,3-propanediol 0 1 1 2-chloroaniline (4,4-methylenebis) regulated carcinogen 2-deoxycytidine 1 0 1 2-dimethylaminoethanol 2-ethoxyethanol 2 2 0 reproductive hazard, skin absorption 2-hydroxyoctanoic acid 2 0 0 2-mercaptonethanol 3 2 1 2-Mercaptoethylamine 201 2-methoxyethanol 121may form peroxides 2-methoxyethyl ether 121may form peroxides 2-methylamino ethanol 320corrosive, poss.sensitizer 2-methyl-3-buten-2-ol 2 3 0 2-methylbutane 1 4 0 2-napthyl methyl ketone 111 2-nitrobenzoic acid 2 0 0 2-nitrofluorene 0 0 1 2-nitrophenol 2 1 1 2-propanol 1 3 0 2-thenoyltrifluoroacetone 1 1 1 2,2-oxydiethanol 1 1 0 2,2,2-tribromoethanol 2 0 -

Potassium Acetate Injection, Solution, Concentrate Hospira, Inc
POTASSIUM ACETATE- potassium acetate injection, solution, concentrate Hospira, Inc. ---------- Potassium Acetate Injection, USP Potassium Acetate Injection, USP 100 mEq/50 mL (2 mEq/mL) + - (2 mEq K /mL and 2 mEq CH3COO /mL) Pharmacy Bulk Package - Not for Direct Infusion. CONCENTRATED SOLUTION – FOR INTRAVENOUS USE ONLY AFTER DILUTION AND THOROUGH MIXING. Glass Fliptop Vial Rx only DESCRIPTION Potassium Acetate Injection, USP (2 mEq/mL) is a sterile, nonpyrogenic, concentrated solution of potassium acetate in water for injection. The solution is administered after dilution by the intravenous route as an electrolyte replenisher. It must not be administered undiluted. Each mL contains 196 mg of potassium acetate which provides 2 mEq each of + potassium (K ) and acetate (CH3COO¯). The solution may contain acetic acid for pH adjustment. The pH is 6.2 (range: 5.5 to 8.0). The osmolar concentration is 4 mOsmol/mL (calc.); specific gravity is 1.089. The Pharmacy Bulk Package is a sterile dosage which contains multiple single doses for use only in a pharmacy bulk admixture program. The solution is intended as an alternative to potassium chloride to provide potassium ion (K+) for addition to large volume infusion fluids for intravenous use. Potassium Acetate, USP is chemically designated CH3COOK, and is comprised of colorless crystals or a white crystalline powder that is very soluble in water. The Pharmacy Bulk Package is designed for use with manual, gravity flow operations and automated compounding devices for preparing sterile parenteral nutrient admixtures. The Pharmacy Bulk Package contains no bacteriostat, antimicrobial agent, or added buffer. Multiple single doses may be dispensed during continual aliquoting operations.