Whole Genome Resequencing Data for Three Rockfish Species of Sebastes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Nursery Origin and Population Connectivity of Swordfish Xiphias Gladius in the North Pacific Ocean
Received: 15 January 2021 Accepted: 9 March 2021 DOI: 10.1111/jfb.14723 REGULAR PAPER FISH Nursery origin and population connectivity of swordfish Xiphias gladius in the North Pacific Ocean R. J. David Wells1,2 | Veronica A. Quesnell1 | Robert L. Humphreys Jr3 | Heidi Dewar4 | Jay R. Rooker1,2 | Jaime Alvarado Bremer1,2 | Owyn E. Snodgrass4 1Department of Marine Biology, Texas A&M University at Galveston, Galveston, Texas Abstract 2Department of Ecology & Conservation Element:Ca ratios in the otolith cores of young-of-the-year (YOY) swordfish, Xiphias Biology, Texas A&M University, College gladius, were used as natural tracers to predict the nursery origin of subadult and adult Station, Texas 3Retired, Pacific Islands Fisheries Science swordfish from three foraging grounds in the North Pacific Ocean (NPO). First, the Center, National Marine Fisheries Service, chemistry of otolith cores (proxy for nursery origin) was used to develop nursery- Honolulu, Hawaii specific elemental signatures in YOY swordfish. Sagittal otoliths of YOY swordfish were 4Southwest Fisheries Science Center, National Marine Fisheries Service, La Jolla, California collected from four regional nurseries in the NPO between 2000 and 2005: (1) Central Equatorial North Pacific Ocean (CENPO), (2) Central North Pacific Ocean (CNPO), Correspondence R. J. David Wells, Texas A&M University at (3) Eastern Equatorial North Pacific Ocean (EENPO) and (4) Western North Pacific Galveston, Department of Marine Biology, Ocean (WNPO). Calcium (43Ca), magnesium (24Mg), strontium (88Sr) and barium (138Ba) 1001 Texas Clipper Rd. Galveston, TX 77554, USA. were quantified in the otolith cores of YOY swordfish using laser ablation inductively Email: [email protected] coupled plasma mass spectrometry. -

Marine Fish Conservation Global Evidence for the Effects of Selected Interventions
Marine Fish Conservation Global evidence for the effects of selected interventions Natasha Taylor, Leo J. Clarke, Khatija Alliji, Chris Barrett, Rosslyn McIntyre, Rebecca0 K. Smith & William J. Sutherland CONSERVATION EVIDENCE SERIES SYNOPSES Marine Fish Conservation Global evidence for the effects of selected interventions Natasha Taylor, Leo J. Clarke, Khatija Alliji, Chris Barrett, Rosslyn McIntyre, Rebecca K. Smith and William J. Sutherland Conservation Evidence Series Synopses 1 Copyright © 2021 William J. Sutherland This work is licensed under a Creative Commons Attribution 4.0 International license (CC BY 4.0). This license allows you to share, copy, distribute and transmit the work; to adapt the work and to make commercial use of the work providing attribution is made to the authors (but not in any way that suggests that they endorse you or your use of the work). Attribution should include the following information: Taylor, N., Clarke, L.J., Alliji, K., Barrett, C., McIntyre, R., Smith, R.K., and Sutherland, W.J. (2021) Marine Fish Conservation: Global Evidence for the Effects of Selected Interventions. Synopses of Conservation Evidence Series. University of Cambridge, Cambridge, UK. Further details about CC BY licenses are available at https://creativecommons.org/licenses/by/4.0/ Cover image: Circling fish in the waters of the Halmahera Sea (Pacific Ocean) off the Raja Ampat Islands, Indonesia, by Leslie Burkhalter. Digital material and resources associated with this synopsis are available at https://www.conservationevidence.com/ -
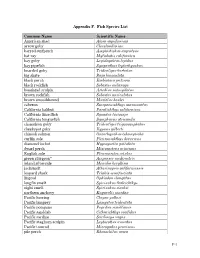
Appendix E: Fish Species List
Appendix F. Fish Species List Common Name Scientific Name American shad Alosa sapidissima arrow goby Clevelandia ios barred surfperch Amphistichus argenteus bat ray Myliobatis californica bay goby Lepidogobius lepidus bay pipefish Syngnathus leptorhynchus bearded goby Tridentiger barbatus big skate Raja binoculata black perch Embiotoca jacksoni black rockfish Sebastes melanops bonehead sculpin Artedius notospilotus brown rockfish Sebastes auriculatus brown smoothhound Mustelus henlei cabezon Scorpaenichthys marmoratus California halibut Paralichthys californicus California lizardfish Synodus lucioceps California tonguefish Symphurus atricauda chameleon goby Tridentiger trigonocephalus cheekspot goby Ilypnus gilberti chinook salmon Oncorhynchus tshawytscha curlfin sole Pleuronichthys decurrens diamond turbot Hypsopsetta guttulata dwarf perch Micrometrus minimus English sole Pleuronectes vetulus green sturgeon* Acipenser medirostris inland silverside Menidia beryllina jacksmelt Atherinopsis californiensis leopard shark Triakis semifasciata lingcod Ophiodon elongatus longfin smelt Spirinchus thaleichthys night smelt Spirinchus starksi northern anchovy Engraulis mordax Pacific herring Clupea pallasi Pacific lamprey Lampetra tridentata Pacific pompano Peprilus simillimus Pacific sanddab Citharichthys sordidus Pacific sardine Sardinops sagax Pacific staghorn sculpin Leptocottus armatus Pacific tomcod Microgadus proximus pile perch Rhacochilus vacca F-1 plainfin midshipman Porichthys notatus rainwater killifish Lucania parva river lamprey Lampetra -

Rockfish (Sebastes) That Are Evolutionarily Isolated Are Also
Biological Conservation 142 (2009) 1787–1796 Contents lists available at ScienceDirect Biological Conservation journal homepage: www.elsevier.com/locate/biocon Rockfish (Sebastes) that are evolutionarily isolated are also large, morphologically distinctive and vulnerable to overfishing Karen Magnuson-Ford a,b, Travis Ingram c, David W. Redding a,b, Arne Ø. Mooers a,b,* a Biological Sciences, Simon Fraser University, 8888 University Drive, Burnaby BC, Canada V5A 1S6 b IRMACS, Simon Fraser University, 8888 University Drive, Burnaby BC, Canada V5A 1S6 c Department of Zoology and Biodiversity Research Centre, University of British Columbia, #2370-6270 University Blvd., Vancouver, Canada V6T 1Z4 article info abstract Article history: In an age of triage, we must prioritize species for conservation effort. Species more isolated on the tree of Received 23 September 2008 life are candidates for increased attention. The rockfish genus Sebastes is speciose (>100 spp.), morpho- Received in revised form 10 March 2009 logically and ecologically diverse and many species are heavily fished. We used a complete Sebastes phy- Accepted 18 March 2009 logeny to calculate a measure of evolutionary isolation for each species and compared this to their Available online 22 April 2009 morphology and imperilment. We found that evolutionarily isolated species in the northeast Pacific are both larger-bodied and, independent of body size, morphologically more distinctive. We examined Keywords: extinction risk within rockfish using a compound measure of each species’ intrinsic vulnerability to Phylogenetic diversity overfishing and categorizing species as commercially fished or not. Evolutionarily isolated species in Extinction risk Conservation priorities the northeast Pacific are more likely to be fished, and, due to their larger sizes and to life history traits Body size such as long lifespan and slow maturation rate, they are also intrinsically more vulnerable to overfishing. -

Osteological Development of Wild-Captured Larvae and a Juvenile Sebastes Koreanus (Pisces, Scorpaenoidei) from the Yellow Sea Hyo Jae Yu and Jin-Koo Kim*
Yu and Kim Fisheries and Aquatic Sciences (2016) 19:20 DOI 10.1186/s41240-016-0021-0 RESEARCH ARTICLE Open Access Osteological development of wild-captured larvae and a juvenile Sebastes koreanus (Pisces, Scorpaenoidei) from the Yellow Sea Hyo Jae Yu and Jin-Koo Kim* Abstract The osteological development in Sebastes koreanus is described and illustrated on the basis of 32 larvae [6.11–11.10 mm body length (BL)] and a single juvenile (18.60 mm BL) collected from the Yellow Sea. The first-ossified skeletal elements, which are related to feeding, swimming, and respiration, appear in larvae of 6.27 mm BL; these include the jaw bones, palatine, opercular, hyoid arch, and pectoral girdle. All skeletal elements are fully ossified in the juvenile observed in the study. Ossification of the neurocranium started in the frontal, pterotic, and parietal regions at 6.27 mm BL, and then in the parasphenoid and basioccipital regions at 8.17 mm BL. The vertebrae had started to ossify at ~7.17 mm BL, and their ossification was nearly complete at 11.10 mm BL. In the juvenile, although ossification of the pectoral girdle was fully complete, the fusion of the scapula and uppermost radial had not yet occurred. Thus, the scapula and uppermost radial fuse during or after the juvenile stage. The five hypurals in the caudal skeleton were also fused to form three hypural elements. The osteological results are discussed from a functional viewpoint and in terms of the comparative osteological development in related species. Keywords: Sebastes koreanus, Korean fish, Larvae, Juvenile, Osteological development Background Sebastes koreanus Kim and Lee 1994, in the family Osteological development in teleost fishes involves a Scorpaenidae (or Sebastidae sensu Nakabo and Kai sequence of remarkable morphological and functional 2013), is smaller than its congeneric species and is changes, occurring in different developmental stages regarded as endemic to the Yellow Sea (Kim and Lee (Löffler et al. -

Management Plan for the Rougheye/Blackspotted Rockfish Complex (Sebastes Aleutianus and S
DRAFT SPECIES AT RISK ACT MANAGESPECIESMENT PLAN AT RISK SERIES ACT MANAGEMENT PLAN SERIES MANAGEMENT PLAN FOR THE ROUGHEYE/BLACKSPOTTEDMANAGEMENT PLAN FOR THE ROCKFISH ROUGHEY E COMPLEXROCKFISHROUGHEYE (SEBASTES ALEUTIANUSROCKFISH COMPLEX AND S. MELANOSTICTUS(SEBASTES ALEUTIANUS) AND LONGSPINE AND S. THORNYHEADMELANOSTICTUS (SEBASTOLOBUS) AND LONGSPINE ALTIVELIS THORNYHE) IN AD CANADA(SEBAST OLOBUS ALTIVELIS)INCANADA SEBASTES ALEUTIANUS; SEBASTES MELANOSTICTUS SEBASTES ASEBASTOLOBUSLEUTIANUS; SEBASTES ALTIVELIS MEL ANOSTICTUS SEBASTOLOBUS ALTIVELIS S. aleutianus S. melanostictus 2012 Photo Credit: DFO Sebastolobus altivelis 2012 About the Species at Risk Act Management Plan Series What is the Species at Risk Act (SARA)? SARA is the Act developed by the federal government as a key contribution to the common national effort to protect and conserve species at risk in Canada. SARA came into force in 2003, and one of its purposes is “to manage species of special concern to prevent them from becoming endangered or threatened.” What is a species of special concern? Under SARA, a species of special concern is a wildlife species that could become threatened or endangered because of a combination of biological characteristics and identified threats. Species of special concern are included in the SARA List of Wildlife Species at Risk. What is a management plan? Under SARA, a management plan is an action-oriented planning document that identifies the conservation activities and land use measures needed to ensure, at a minimum, that a species of special concern does not become threatened or endangered. For many species, the ultimate aim of the management plan will be to alleviate human threats and remove the species from the List of Wildlife Species at Risk. -

Southward Range Extension of the Goldeye Rockfish, Sebastes
Acta Ichthyologica et Piscatoria 51(2), 2021, 153–158 | DOI 10.3897/aiep.51.68832 Southward range extension of the goldeye rockfish, Sebastes thompsoni (Actinopterygii: Scorpaeniformes: Scorpaenidae), to northern Taiwan Tak-Kei CHOU1, Chi-Ngai TANG2 1 Department of Oceanography, National Sun Yat-sen University, Kaohsiung, Taiwan 2 Department of Aquaculture, National Taiwan Ocean University, Keelung, Taiwan http://zoobank.org/5F8F5772-5989-4FBA-A9D9-B8BD3D9970A6 Corresponding author: Tak-Kei Chou ([email protected]) Academic editor: Ronald Fricke ♦ Received 18 May 2021 ♦ Accepted 7 June 2021 ♦ Published 12 July 2021 Citation: Chou T-K, Tang C-N (2021) Southward range extension of the goldeye rockfish, Sebastes thompsoni (Actinopterygii: Scorpaeniformes: Scorpaenidae), to northern Taiwan. Acta Ichthyologica et Piscatoria 51(2): 153–158. https://doi.org/10.3897/ aiep.51.68832 Abstract The goldeye rockfish,Sebastes thompsoni (Jordan et Hubbs, 1925), is known as a typical cold-water species, occurring from southern Hokkaido to Kagoshima. In the presently reported study, a specimen was collected from the local fishery catch off Keelung, northern Taiwan, which represents the first specimen-based record of the genus in Taiwan. Moreover, the new record ofSebastes thompsoni in Taiwan represented the southernmost distribution of the cold-water genus Sebastes in the Northern Hemisphere. Keywords cold-water fish, DNA barcoding, neighbor-joining, new recorded genus, phylogeny, Sebastes joyneri Introduction On an occasional survey in a local fish market (25°7.77′N, 121°44.47′E), a mature female individual of The rockfish genusSebastes Cuvier, 1829 is the most spe- Sebastes thompsoni (Jordan et Hubbs, 1925) was obtained ciose group of the Scorpaenidae, which comprises about in the local catches, which were caught off Keelung, north- 110 species worldwide (Li et al. -

Historical Fish Specimens Collected from the Tohoku District by the Saito Ho-On Kai Museum of Natural History
Bull. Natl. Mus. Nat. Sci., Ser. A, 35(1), pp. 9–54, March 22, 2009 Historical Fish Specimens Collected from the Tohoku District by the Saito Ho-on Kai Museum of Natural History Keiichi Matsuura1, Gento Shinohara2 and Masanori Nakae1 1 Collection Center, National Museum of Nature and Science, 3–23–1 Hyakunin-cho, Shinjuku-ku, Tokyo, 169–0073 Japan E-mail: [email protected]; [email protected] 2 Department of Zoology, National Museum of Nature and Science, 3–23–1 Hyakunin-cho, Shinjuku-ku, Tokyo, 169–0073 Japan E-mail: [email protected] Abstract The fish collection of the Saito Ho-on Kai Museum of Natural History was transferred to the National Museum of Nature and Science, Tokyo in February 2006. Ninety percent of the fish collection contains specimens collected from the Tohoku District during the period from 1930 to 1933 when natural environments of Japan were in good condition for various groups of fishes. The fish specimens from the Tohoku District were classified into 361 species/subspecies of 273 genera belonging to 131 families of 31 orders. A list of the species is shown with remarks on distribution. Key words: Fish specimens, Saito Ho-on Kai Museum, Tohoku District, inventory. stead of natural sicence. The museum has tried to Introduction keep its activity at the level before the war, but it The Saito Ho-on Kai Museum was established failed to do so because of financial difficulties. In in November 1933 in Sendai City, Miyagi Pre- 2005, the Saito Ho-on Kai Museum of Natural fecture, Japan. -

Periodic Status Review for the Steller Sea Lion
STATE OF WASHINGTON January 2015 Periodic Status Review for the Steller Sea Lion Gary J. Wiles The Washington Department of Fish and Wildlife maintains a list of endangered, threatened, and sensitive species (Washington Administrative Codes 232-12-014 and 232-12-011, Appendix E). In 1990, the Washington Wildlife Commission adopted listing procedures developed by a group of citizens, interest groups, and state and federal agencies (Washington Administrative Code 232-12-297, Appendix A). The procedures include how species listings will be initiated, criteria for listing and delisting, a requirement for public review, the development of recovery or management plans, and the periodic review of listed species. The Washington Department of Fish and Wildlife is directed to conduct reviews of each endangered, threatened, or sensitive wildlife species at least every five years after the date of its listing. The reviews are designed to include an update of the species status report to determine whether the status of the species warrants its current listing status or deserves reclassification. The agency notifies the general public and specific parties who have expressed their interest to the Department of the periodic status review at least one year prior to the five-year period so that they may submit new scientific data to be included in the review. The agency notifies the public of its recommendation at least 30 days prior to presenting the findings to the Fish and Wildlife Commission. In addition, if the agency determines that new information suggests that the classification of a species should be changed from its present state, the agency prepares documents to determine the environmental consequences of adopting the recommendations pursuant to requirements of the State Environmental Policy Act. -

Pacific False Kelpfish, Sebastiscus Marmoratus (Cuvier, 1829) (Scorpaeniformes, Sebastidae) Found in Norwegian Waters
BioInvasions Records (2018) Volume 7, Issue 1: 73–78 Open Access DOI: https://doi.org/10.3391/bir.2018.7.1.11 © 2018 The Author(s). Journal compilation © 2018 REABIC Research Article Pacific false kelpfish, Sebastiscus marmoratus (Cuvier, 1829) (Scorpaeniformes, Sebastidae) found in Norwegian waters Haakon Hansen1,* and Egil Karlsbakk2 1Norwegian Veterinary Institute, P.O. Box 750 Sentrum, NO-0106 Oslo, Norway 2Department of Biology, University of Bergen, 5020 Bergen, Norway Author e-mails: [email protected] (HH), [email protected] (EK) *Corresponding author Received: 28 September 2017 / Accepted: 21 November 2017 / Published online: 1 December 2017 Handling editor: Henn Ojaveer Abstract During an angling competition in the Oslofjord, Southern Norway, a fish species previously unknown to the anglers was caught. Subsequent morphological studies and DNA barcoding identified it as a false kelpfish, Sebastiscus marmoratus (Cuvier, 1829), a species native to the Western Pacific from southern Hokkaido, Japan to the Philippines. The specimen was a female with a length of 29.2 cm and weighing 453 g. Stomach contents revealed fish remains, as well as the brachyuran Xantho pilipes A. Milne-Edwards, 1867 and remains of anomuran decapods. Parasitological examination revealed infections with the locally common generalist parasites Derogenes varicus (Digenea) and Hysterothylacium aduncum (Nematoda) that likely have been acquired through prey fish. A literature study of the parasites of S. marmoratus was carried out, listing at least 31 species. To the best of our knowledge, this is the first report of this fish species in the Atlantic. The introduction route is unknown, but the most likely possibility is via a ship’s ballast water as a larva or fry, which again would imply that the specimen has been in temperate waters for several years. -

Venom Evolution Widespread in Fishes: a Phylogenetic Road Map for the Bioprospecting of Piscine Venoms
Journal of Heredity 2006:97(3):206–217 ª The American Genetic Association. 2006. All rights reserved. doi:10.1093/jhered/esj034 For permissions, please email: [email protected]. Advance Access publication June 1, 2006 Venom Evolution Widespread in Fishes: A Phylogenetic Road Map for the Bioprospecting of Piscine Venoms WILLIAM LEO SMITH AND WARD C. WHEELER From the Department of Ecology, Evolution, and Environmental Biology, Columbia University, 1200 Amsterdam Avenue, New York, NY 10027 (Leo Smith); Division of Vertebrate Zoology (Ichthyology), American Museum of Natural History, Central Park West at 79th Street, New York, NY 10024-5192 (Leo Smith); and Division of Invertebrate Zoology, American Museum of Natural History, Central Park West at 79th Street, New York, NY 10024-5192 (Wheeler). Address correspondence to W. L. Smith at the address above, or e-mail: [email protected]. Abstract Knowledge of evolutionary relationships or phylogeny allows for effective predictions about the unstudied characteristics of species. These include the presence and biological activity of an organism’s venoms. To date, most venom bioprospecting has focused on snakes, resulting in six stroke and cancer treatment drugs that are nearing U.S. Food and Drug Administration review. Fishes, however, with thousands of venoms, represent an untapped resource of natural products. The first step in- volved in the efficient bioprospecting of these compounds is a phylogeny of venomous fishes. Here, we show the results of such an analysis and provide the first explicit suborder-level phylogeny for spiny-rayed fishes. The results, based on ;1.1 million aligned base pairs, suggest that, in contrast to previous estimates of 200 venomous fishes, .1,200 fishes in 12 clades should be presumed venomous. -

(Sebastes Chrysomelas) from Central California
Fall 2013 237 California Fish and Game 99(4):237-239; 2013 Orange coloration in a black-and-yellow rockfish (Sebastes chrysomelas) from central California KEVIN O. LEWAND, JOHN R. HYDE, VINCE P. BUONACCORSI, AND ROBERT N. LEA Monterey Bay Aquarium, 886 Cannery Row, Monterey, CA 93940, USA (KOL) Southwest Fisheries Science Center,8604 La Jolla Shores Drive, La Jolla, CA 92037-1508, USA (JRH) Von Liebig Science Center, 601 17th Street, Huntingdon, PA 16652, USA (VPB) California Department of Fish and Game (retired), 20 Lower Ragsdale Drive, Monterey, CA 93940, USA and California Academy of Sciences, 55 Music Concourse Drive, Golden Gate Park, San Francisco, CA 94118, USA (RNL) *Correspondent: [email protected] Key words: abnormal coloration, black-and-yellow rockfish,Sebastes chrysomelas ________________________________________________________________________ In November 2010 an orange colored rockfish (Sebastes sp.) was caught by hook and line off Avila Beach, San Luis Obispo County, California (35º 14’ N, 120º 64’ W) at a depth of 6 m near the Point San Luis Lighthouse. The fish initially was not identifiable with any of the shallow-water rockfishes, yet had the general conformation of a member of the Pteropodus rockfish complex. Through analysis of morphological characters the specimen was determined to be either a black-and-yellow rockfish (S. chrysomelas) or gopher rockfish (S. carnatus). Both are considered as shallow-water species with gopher rockfish generally found deeper than black-and-yellow rockfish (Larson 1980, Love et al. 2002) and we initially assumed that the orange colored rockfish was a black-and-yellow rockfish. Within aquarium conditions, the fish demonstrated behavioral characteristics similar to those of both black-and- yellow rockfish and gopher rockfish by hiding in cracks in rocks covered with invertebrates such as California hydrocoral (Stylaster californicus), strawberry anemone (Corynactis californica), sponges (Porifera), and algae (Figure 1).