Pleuronectiformes) to Boost Aquaculture Production
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
2018 Final LOFF W/ Ref and Detailed Info
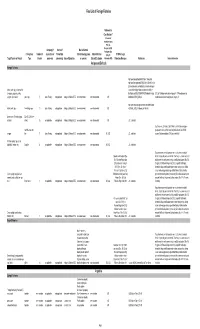
Final List of Foreign Fisheries Rationale for Classification ** (Presence of mortality or injury (P/A), Co- Occurrence (C/O), Company (if Source of Marine Mammal Analogous Gear Fishery/Gear Number of aquaculture or Product (for Interactions (by group Marine Mammal (A/G), No RFMO or Legal Target Species or Product Type Vessels processor) processing) Area of Operation or species) Bycatch Estimates Information (N/I)) Protection Measures References Detailed Information Antigua and Barbuda Exempt Fisheries http://www.fao.org/fi/oldsite/FCP/en/ATG/body.htm http://www.fao.org/docrep/006/y5402e/y5402e06.htm,ht tp://www.tradeboss.com/default.cgi/action/viewcompan lobster, rock, spiny, demersal fish ies/searchterm/spiny+lobster/searchtermcondition/1/ , (snappers, groupers, grunts, ftp://ftp.fao.org/fi/DOCUMENT/IPOAS/national/Antigua U.S. LoF Caribbean spiny lobster trap/ pot >197 None documented, surgeonfish), flounder pots, traps 74 Lewis Fishing not applicable Antigua & Barbuda EEZ none documented none documented A/G AndBarbuda/NPOA_IUU.pdf Caribbean mixed species trap/pot are category III http://www.nmfs.noaa.gov/pr/interactions/fisheries/tabl lobster, rock, spiny free diving, loops 19 Lewis Fishing not applicable Antigua & Barbuda EEZ none documented none documented A/G e2/Atlantic_GOM_Caribbean_shellfish.html Queen conch (Strombus gigas), Dive (SCUBA & free molluscs diving) 25 not applicable not applicable Antigua & Barbuda EEZ none documented none documented A/G U.S. trade data Southeastern U.S. Atlantic, Gulf of Mexico, and Caribbean snapper- handline, hook and grouper and other reef fish bottom longline/hook-and-line/ >5,000 snapper line 71 Lewis Fishing not applicable Antigua & Barbuda EEZ none documented none documented N/I, A/G U.S. -

Effects of Acute and Chronic Air Exposure on Growth and Stress Response of Juvenile Olive Flounder, Paralichthys Olivaceus
www.trjfas.org ISSN 1303-2712 Turkish Journal of Fisheries and Aquatic Sciences 18:143-151 (2018) DOI: 10.4194/1303-2712-v18_1_16 RESEARCH PAPER Effects of Acute and Chronic Air Exposure on Growth and Stress Response of Juvenile Olive Flounder, Paralichthys olivaceus Han Kyu Lim1, Jun Wook Hur2,* 1Mokpo National University, Marine and Fisheries Resources, 1666 Youngsan-ro, Cheonggye, Muan, Jeonnam 58554, Korea. 2 Bio-Monitoring Center, 202 ho, 49, 1730 Beon-gil, Dongseodae-ro, Dong-gu, Daejeon, 300-805, Korea. * Corresponding Author: Tel.: +82.42 6386845; Fax: +82.42 6386845; Received 10 September 2016 E-mail: [email protected] Accepted 23 May 2017 Abstract We studied the effects of acute and chronic exposure to air on the growth and stress response of juvenile olive flounder, Paralichthys olivaceus. To study the stress response, the water was completely drained from the experimental tank, and the stressed group was exposed to air for 5 minutes, after which the tank was refilled with water. This stress was repeated daily for 30 days (between 1200 and 1300 h). From day 31 to day 69, no stress was applied. On day 70, the fish were again exposed to the air. The non-stressed group was not subjected to air exposure during the 70 days. We measured cortisol, glucose and lactic acid levels, osmolality, growth, survival, and feeding responses during the 70-day test period. Our results showed that olive flounder exhibit “typical” physiological responses (in cortisol, glucose, and lactic acid levels and osmolality) to the acute stress induced by air exposure. The response to chronic stress showed a similar increasing tendency. -

Recycled Fish Sculpture (.PDF)
Recycled Fish Sculpture Name:__________ Fish: are a paraphyletic group of organisms that consist of all gill-bearing aquatic vertebrate animals that lack limbs with digits. At 32,000 species, fish exhibit greater species diversity than any other group of vertebrates. Sculpture: is three-dimensional artwork created by shaping or combining hard materials—typically stone such as marble—or metal, glass, or wood. Softer ("plastic") materials can also be used, such as clay, textiles, plastics, polymers and softer metals. They may be assembled such as by welding or gluing or by firing, molded or cast. Researched Photo Source: Alaskan Rainbow STEP ONE: CHOOSE one fish from the attached Fish Names list. Trout STEP TWO: RESEARCH on-line and complete the attached K/U Fish Research Sheet. STEP THREE: DRAW 3 conceptual sketches with colour pencil crayons of possible visual images that represent your researched fish. STEP FOUR: Once your fish designs are approved by the teacher, DRAW a representational outline of your fish on the 18 x24 and then add VALUE and COLOUR . CONSIDER: Individual shapes and forms for the various parts you will cut out of recycled pop aluminum cans (such as individual scales, gills, fins etc.) STEP FIVE: CUT OUT using scissors the various individual sections of your chosen fish from recycled pop aluminum cans. OVERLAY them on top of your 18 x 24 Representational Outline 18 x 24 Drawing representational drawing to judge the shape and size of each piece. STEP SIX: Once you have cut out all your shapes and forms, GLUE the various pieces together with a glue gun. -

Histological Observation on Adult Gonads from Meiogynogentic Olive Flounder Paralichthys Olivaceus
INTERNATIONAL JOURNAL OF AGRICULTURE & BIOLOGY ISSN Print: 1560–8530; ISSN Online: 1814–9596 17F–136/2018/20–3–689–694 DOI: 10.17957/IJAB/15.0562 http://www.fspublishers.org Full Length Article Histological Observation on Adult Gonads from Meiogynogentic Olive Flounder Paralichthys olivaceus Deyou Ma1,3, Shenda Weng2, Peng Sun5, Jun Li2,4, Peijun Zhang2 and Feng You2,4* 1Key Laboratory of Mariculture & Stock Enhancement in North China, Ministry of Agriculture, Dalian Ocean University, Dalian-116023, China 2Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Chinese Academy of Sciences, Qingdao-266071, China 3Key laboratory of Fish Applied Biology and Aquaculture in North China, Liaoning Province, Dalian Ocean University, Dalian-116023, China 4Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao-266071, China 5Key Laboratory of East China Sea and Oceanic Fishery Resources Exploitation, Ministry of Agriculture, East China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Shanghai-200090, China *For correspondence: [email protected] Abstract Gynogenesis is a common method to manipulate chromosomes of aquaculture animals with sex dimorphism. The adverse effects on gonad development can be identified through morphology and histology. The goal of this study was to examine the gonadal development of twenty-three meiogynogenetic olive flounder Paralichthys olivaceus samples of two-years age using histological and immunohistochemical methods. We found that ovaries of nine individuals developed normally, while those of fourteen fish exhibited some distinct malformation, including a pair of asymmetrically developed lobes (divided into only one lobe and one slowly developed lobe) and a pair of slowly developed lobes. -

Screening of the White Margined Sole, Synaptura Marginata (Soleidae), As a Candidate for Aquaculture in South Africa
Screening of the white margined sole, Synaptura marginata (Soleidae), as a candidate for aquaculture in South Africa THESIS Submitted in fulfilment of the requirements for the degree of MASTER OF SCIENCE Department of Ichthyology and Fisheries Science Rhodes University, Grahamstown South Africa By Ernst Frederick Thompson September 2003 The white-margined sole, Synaptura marginata (Boulenger, 1900)(Soleidae), 300 mm TL (Kleinemonde). Photograph: James Stapley Table of Contents Abstract Acknowledgements Chapter 1 - General Introduction .. .......... ............ .. .... ......... .. .. ........ 1 Chapter 2 - General Materials and Methods .................................... 12 Chapter 3 - Age and Growth Introduction ................................. .. ................ .. ............ ... .. 19 Materials and Methods .................. ... ... .. .. .............. ... ........... 21 Results ........... ... ............. .. ....... ............ .. .... ... ................... 25 Discussion .......................................... .. ................ ..... ....... 37 Chapter 4 - Feeding Biology Introduction ................................... .......... ........................ .40 Materials and Methods ............................................. ... ...... .43 Results ................................................... ....................... .47 Discussion .. .................... ........... .. .... .. .......... ...... ............. .49 Chapter 5 - Reproduction Introduction ........................ ... ......... ......... ........ -

Microsatellite Analysis As a Tool for Discriminating an Interfamily Hybrid Between Olive Flounder and Starry Flounder
Microsatellite analysis as a tool for discriminating an interfamily hybrid between olive flounder and starry flounder J.-H. Kang1, Y.-K. Kim1, J.-Y. Park1, C.-M. An1, M.-M. Nam2, S.G. Byun2, B.I. Lee2, J.H. Lee2 and T.-J. Choi3 1Biotechnology Research Division, Busan, Korea 2East Sea Fisheries Research Institute, Uljin, Korea 3Department of Microbiology, Pukyong National University, Busan, Korea Corresponding author: T.-J. Choi E-mail: [email protected] Genet. Mol. Res. 10 (4): 2786-2794 (2011) Received May 30, 2011 Accepted September 7, 2011 Published October 31, 2011 DOI http://dx.doi.org/10.4238/2011.October.31.16 ABSTRACT. An interspecific artificial hybrid was produced between two economically important aquaculture flatfish: olive flounder (Paralichthys olivaceus) and starry flounder (P. stellatus). This hybrid displays the rapid growth characteristic of the former and tolerance to low temperatures and low salinity of the latter, but the genetics of inheritance in this hybrid have not been elucidated. Polymorphic microsatellite markers developed for P. olivaceus and P. stellatus were tested to determine if these markers can be used for analysis of parentage and genetic inheritance. Multiplex PCR using two primer sets that were specific to each species produced PCR products of different sizes; these could be used for the identification of interspecific hybrids. Among the 192 primers derived from olive flounder, 25.5% of the primer sets successfully amplified genomic DNA from starry flounder, and 23% of the 56 primer sets originating from starry flounder amplified DNA from olive flounder. Analysis of genetic inheritance in the hybrid using seven of the 62 microsatellite markers common to both species demonstrated classic Mendelian inheritance of these markers in the hybrid progeny, with the exception of one locus identified as a null allele in the hybrid. -

The United States and Japanese Counterpart Panels on Aquaculture
PREFACE TheUnited States and Japanese counterpart panels onaquaculture wereformed in 1969 under the UnitedStates-Japan Cooperative Program inNatural Resources UJNR! The panels currently include specialistsdrawnfrom the government andacadeinic departinents mostconcerned withaquaculture- Chargedwith exploring anddeveloping bilateral cooperation, thepanels have focused their efforts on exchanginginformation related to aquaculture thatcould be of benefittoboth countries. TheUJNR was begun during theThird Cabinet Level Meeting ofthe Joint United States-Japan CommitteeonTrade and Economic Affairsin January 1964,ln addition toaquaculture, currentsubjects in theprogram include toxicmicroorganisms, airpollution, energy, forage crops, national parkmanagement, rnycoplasmosis,windand seismic effects, protein resources, forestry, andseveral joint panels and corninitteesin marine resolve research, development, andutilization. Accomplishmentsiticlude:increased communication andcooperation amongtechnical specialists; exchangesofinformation, data,and research findings; annual meetings ofthe panel, a policy-coordinating body;administrative staffmeetings; exchanges ofequipment, materials, andsamples; several major technicalconferences; andbeneficial effects of internationalrelations. The26th U.S.-Japan Aquaculture PanelSyinposium washeld in Durham, New Hampshire, from16- 18September ] 997. Following thesyrnposiuin, fieldtrips during a seven-day period included theareas of Portsmouth,NewHainpshire; andBar Harbor, Eastport, Camden, and Boothbay Harbor, Maine. -

General Distribution OCDE/GD(97)119
General Distribution OCDE/GD(97)119 TOWARDS SUSTAINABLE FISHERIES: COUNTRY REPORTS The OECD study Towards Sustainable Fisheries is composed of 3 volumes. The publication on sale "Towards Sustainable Fisheries: Economic Aspects of the Management of Living Marine Resources" provides a comprehensive assessment of the economic performance of management regimes based on the analysis of over 100 fisheries in the OECD countries. The general diffusion document "Towards Sustainable Fisheries: Issues Papers", contains thematic contributions which were prepared for the project by individual countries based on their experiences. Finally, the Country Reports which are contained in this volume describe national fishery management policies in Member countries. ORGANISATION FOR ECONOMIC CO-OPERATION AND DEVELOPMENT Paris 54740 Document complet disponible sur OLIS dans son format d'origine Complete document available on OLIS in its original format Applications for permission to reproduce or translate all or part of this material should be made to: Head of Publications Service, OECD, 2, rue André-Pascal, 75775 Paris Cedex 16, France. Copyright OECD, 1997. 2 FOREWORD The OECD study Towards Sustainable Fisheries is composed of 3 volumes. The publication on sale Towards Sustainable Fisheries: Economic Aspects of the Management of Living Marine Resources provides a comprehensive assessment of the economic performance of management regimes based on the analysis of over 100 fisheries in the OECD countries. The general diffusion document Towards Sustainable Fisheries: Issue Papers contains thematic contributions which were prepared for the project by individual countries based on their experiences. Finally, the Country Reports which are contained in this volume describe national fishery management policies in Member countries. -

Effect of Hyposalinity on the Infection and Pathogenicity of Miamiensis Avidus Causing Scutic- Ociliatosis in Olive Flounder Paralichthys Olivaceus
Vol. 86: 175–179, 2009 DISEASES OF AQUATIC ORGANISMS Published September 23 doi: 10.3354/dao02116 Dis Aquat Org NOTE Effect of hyposalinity on the infection and pathogenicity of Miamiensis avidus causing scutic- ociliatosis in olive flounder Paralichthys olivaceus Nanae Takagishi, Tomoyoshi Yoshinaga*, Kazuo Ogawa Department of Aquatic Bioscience, Graduate School of Agricultural and Life Sciences, University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan ABSTRACT: Miamiensis avidus, a causative agent of scuticociliatosis in olive flounder Paralichthys olivaceus, was previously reported to proliferate the fastest in media with an osmolarity of 300 to 500 mOsm kg–1. This suggests that hyposaline conditions can promote the development of the dis- ease. In the present study, olive flounder constantly showed high mortalities when they were exper- imentally challenged with the parasite by immersion and subsequently reared in hyposaline condi- tions. Furthermore, affected flounder produced by the challenge showed symptoms identical to those in naturally infected flounder. It was experimentally demonstrated that hyposaline conditions can be a key factor for the development and outbreak of scuticociliatosis in olive flounder. KEY WORDS: Hyposaline condition · Immersion challenge · Miamiensis avidus · Scuticociliate Resale or republication not permitted without written consent of the publisher INTRODUCTION The occurrences of scuticociliatosis seem to be influ- enced by environmental and fish conditions. However, Scuticociliates infect aquatic organisms opportunisti- these conditions have not been specified yet. Based on cally. Outbreaks of scuticociliate infection occur in anecdotal evidence, olive flounder suffer from scutic- many fish species, including olive flounder Par- ociliatosis more frequently in land-based aquaculture alichthys olivaceus (Yoshinaga & Nakazoe 1993, Kim facilities supplied with water from saltwater wells in et al. -
Draft List of Foreign Fisheries
Classification ** (Presence of mortality or injury (P/A), Co- Company (if Marine Mammal Occurrence (C/O), Fishery/Gear Number of aquaculture or Interactions (by group Marine Mammal Analogous Gear Target Species or Product Type Vessels processor) Area of Operation or species) Bycatch Estimates (A/G), No Antigua and Barbuda Exempt Fisheries lobster, rock, spiny, demersal fish (snappers, groupers, grunts, surgeonfish), flounder pots, traps 74 Lewis Fishing Antigua & Barbuda EEZ none documented none documented A/G lobster, rock, spiny free diving, loops 19 Lewis Fishing Antigua & Barbuda EEZ none documented none documented A/G Queen conch (Strombus gigas), Dive (SCUBA & free molluscs diving) 25 not applicable Antigua & Barbuda EEZ none documented none documented A/G handline, hook and snapper line 71 Lewis Fishing Antigua & Barbuda EEZ none documented none documented N/I, A/G Off-shore pelagic spp. (tunas, dolphinfish, wahoo, etc.) Drop line 6 not applicable Antigua & Barbuda EEZ none documented none documented N/I, A/G Coastal pelagic spp. (southern sennets, jacks, mullets, herrings, etc.) Beach seine 1 not applicable Antigua & Barbuda EEZ none documented none documented N/I, A/G Coastal pelagic spp. (herrings, ballyhoo, etc.) Bait net 1 not applicable Antigua & Barbuda EEZ none documented none documented N/I, A/G Export Fisheries Marine mammals susceptible to gillnet gear in coastal waters of the Caribbean: tucuxi, pygmy sperm whale, Risso's dolphin, bottlenose dolphin, Atlantic spotted Demersal spp. (grunts, parrotfish, dolphin, killer -

Platichthys Stellatus): a Potential New Candidate for Aquaculture in Temperate Regions J
11(3): 017-025(2017) Journal of FisheriesSciences.com E-ISSN 1307-234X © 2017 www.fisheriessciences.com Research Article Effect of Beta-1-3-Glucan and Mannans on Growth and Fitness of Starry Flounder (Platichthys Stellatus): A Potential New Candidate for Aquaculture in Temperate Regions J. Schmidt1*, A.A. Bischoff2, M. Weiß1, S.K. Kim3, S. Frickenhaus1,4, M.J. Slater1 and B.H. Buck1,4 1Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research (AWI), Am Handelshafen, Bremerhaven, Germany 2University of Rostock, Justus-von-Liebig Weg, Rostock, Germany 3West Sea Fisheries Research Institute (NFRDI), Junggu Ulwangdong, Incheon, Korea 4Bremerhaven University of Applied Sciences, Biotechnology, An der Karlstadt, Bremerhaven, Germany Received: 03.06.2017 / Accepted: 14.06.2017 / Published: 17.06.2017 Abstract: Continuously intensifying aquaculture demands reductions in pathogen infections without increased therapeutics use. A potential solution is the use of prebiotic feed additives like β-glucan and mannan oligosaccharides (MOS). This study focusses (1) on the effect of prebiotics glucan/MOS on growth and fitness of Starry flounder and (2) on the viability of Starry flounder as an aquaculture candidate (as it is considered in South Korea). Over 56- days, juvenile Starry flounder were fed with glucan/MOS enhanced diet and a control diet. Feeding behavior, growth rate, morphological and blood physiological parameters were monitored. Fish fed glucan/MOS enriched diets exhibited significantly increased growth over the experimental period (GLM, p<0.01). Concentrations of cholesterol (P=0.043) and albumin (P=0.016) were significantly increased in the blood plasma of fish fed glucan/MOS. Whole body proximate analysis revealed significantly elevated crude protein (P=<0.001) and lipid (P=<0.005) in fish fed glucan/MOS compared to the control. -

No. Name Poster Title Student
No. Name Poster Title Student 1 Landis, Eric Aquaculture Drugs: What Are They And Why Does Fda Review Matter? No 2 Crosby, Tina Epidemiological Cutoff Value Analysis Of 229 Mics From Standard Testing Of Flavobacterium Columnare Isolates By Four Laboratories No 3 Ulloa, Pilar Effect Of Β-Glucans On Intestinal Health In Zebrafish And Yellowtail Kingfish No 4 Soto-Davila, Manuel Antibacterial Effects Of Cholecalciferol In Atlantic Salmon (Salmo Salar) Primary Macrophages Infected With Aeromonas Salmonicida. Yes 5 Kang, So Young Efficacy Of Optimized Rhus Verniciflua Stokes Extracts Against Edwardsiellosis In Experimental And Field Trials In Olive Flounder Paralichthys Olivaceus No 6 Soto, Esteban Pharmacokinetic Parameters Following A Sinlge Intravenous Administration Of 10 Mg/Kg Of Natamycin To White Sturgeon (Acipenser Transmontanus) No 7 Jung-Schroers, Verena Development Of Antibiotic Resistances In Bacteria Isolated From Ornamental Fish And Fish For Food Production Between 2005 And 2017 No Diagnostic Methods For The Identification Of Different Aeromonas Spp. And Examination Of Their Pathogenicity Factors And Their Cytotoxicity And Adherence To 8 Jung-Schroers, Verena Fish Mucus No 9 Jung-Schroers, Verena Identification And Pathogenicity Of Vibrio Spp. From Recirculating Aquaculture Systems For Pacific White Shrimps (Litopennaeus Vannamei) No 10 Jung-Schroers, Verena Methods For Identification And Differentiation Of Shewanella Spp. Isolates For Diagnostic Use No 11 Ghosh, Satyaki Molecular Detection And Quantification Of The Fish Pathogen Saprolegnia Using Qpcr And Loop Mediated Isothermal Amplification (Lamp). Yes 12 Soto, Esteban Investigating The Role Of The Type Vi Secretion System (T6ss) In The Emergent Fish Pathogen Francisella Noatunensis Subsp. Orientalis ( Changed to Oral ) No 13 Weatherhead, Rebekah The Influence Of Water Temperature On Juvenile Nile Tilapia (Oreochromis Niloticus) Gene Expression Patterns When Challenged With Saprolegnia Parasitica Yes 14 Sebastião, Fernanda Molecular Detection Of Francisella Noatunensis Subsp.