Itga3 Axis Is a Key Regulator of Prosensory Area During Early Inner Ear Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Molecular and Genetic Analysis of Otosclerosis
A molecular and genetic analysis of otosclerosis Joanna Lauren Ziff Submitted for the degree of PhD University College London January 2014 1 Declaration I, Joanna Ziff, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Where work has been conducted by other members of our laboratory, this has been indicated by an appropriate reference. 2 Abstract Otosclerosis is a common form of conductive hearing loss. It is characterised by abnormal bone remodelling within the otic capsule, leading to formation of sclerotic lesions of the temporal bone. Encroachment of these lesions on to the footplate of the stapes in the middle ear leads to stapes fixation and subsequent conductive hearing loss. The hereditary nature of otosclerosis has long been recognised due to its recurrence within families, but its genetic aetiology is yet to be characterised. Although many familial linkage studies and candidate gene association studies to investigate the genetic nature of otosclerosis have been performed in recent years, progress in identifying disease causing genes has been slow. This is largely due to the highly heterogeneous nature of this condition. The research presented in this thesis examines the molecular and genetic basis of otosclerosis using two next generation sequencing technologies; RNA-sequencing and Whole Exome Sequencing. RNA–sequencing has provided human stapes transcriptomes for healthy and diseased stapes, and in combination with pathway analysis has helped identify genes and molecular processes dysregulated in otosclerotic tissue. Whole Exome Sequencing has been employed to investigate rare variants that segregate with otosclerosis in affected families, and has been followed by a variant filtering strategy, which has prioritised genes found to be dysregulated during RNA-sequencing. -

Mir-96 Regulates the Progression of Differentiation in Mammalian Cochlear Inner and Outer Hair Cells
miR-96 regulates the progression of differentiation in mammalian cochlear inner and outer hair cells Stephanie Kuhna,1, Stuart L. Johnsona,1, David N. Furnessb, Jing Chenc, Neil Inghamc, Jennifer M. Hiltonc, Georg Steffesc, Morag A. Lewisc, Valeria Zampinia,d, Carole M. Hackneya, Sergio Masettod, Matthew C. Holleya, Karen P. Steelc, and Walter Marcottia,2 aDepartment of Biomedical Science, University of Sheffield, Sheffield S10 2TN, United Kingdom; bInstitute for Science and Technology in Medicine, Keele University, Keele ST5 5BG, United Kingdom; cWellcome Trust Sanger Institute, Hinxton, Cambridge CB10 1SA, United Kingdom; and dDepartment of Physiology, University of Pavia, 27100 Pavia, Italy Edited by Mary-Claire King, University of Washington, Seattle, WA, and approved December 27, 2010 (received for review November 8, 2010) MicroRNAs (miRNAs) are small noncoding RNAs able to regulate cells up to at least P5 (23) and in the spiral ganglion up to P14 a broad range of protein-coding genes involved in many biological (19). Mutations in miR-96 have been associated with non- processes. miR-96 is a sensory organ-specific miRNA expressed in syndromic progressive hearing loss in humans (15) and mice (23). the mammalian cochlea during development. Mutations in miR-96 We found that hair cell development in diminuendo mice with cause nonsyndromic progressive hearing loss in humans and mice. a Mir96 mutation is arrested at around the day of birth, a time The mouse mutant diminuendo has a single base change in the when IHCs and OHCs still exhibit qualitatively similar bio- seed region of the Mir96 gene leading to widespread changes in physical properties. -

Microrna-1182 and Let-7A Exert Synergistic Inhibition on Invasion
Pan et al. Cancer Cell Int (2021) 21:161 https://doi.org/10.1186/s12935-021-01797-z Cancer Cell International PRIMARY RESEARCH Open Access MicroRNA-1182 and let-7a exert synergistic inhibition on invasion, migration and autophagy of cholangiocarcinoma cells through down-regulation of NUAK1 Xin Pan*, Gang Wang and Baoming Wang Abstract Background: Cholangiocarcinoma (CCA) is the second most common primary liver malignancy worldwide. Several microRNAs (miRNAs) have been implicated as potential tumor suppressors in CCA. This study aims to explore the potential efects of miR-1182 and let-7a on CCA development. Methods: Bioinformatics analysis was conducted to screen diferentially expressed genes in CCA, Western blot analysis detected NUAK1 protein expression and RT-qPCR detected miR-1182, let-7a and NUAK1 expression in CCA tissues and cell lines. Dual luciferase reporter gene assay and RIP were applied to validate the relationship between miR-1182 and NUAK1 as well as between let-7a and NUAK1. Functional experiment was conducted to investigate the role of miR-1182, let-7a and NUAK1 in cell migration, proliferation and autophagy. Then, the CCA cells that received various treatments were implanted to mice to establish animal model, followed by tumor observation and HE staining to evaluate lung metastasis. Results: CCA tissues and cells were observed to have a high expression of NUAK1 and poor expression of miR-1182 and let-7a. NUAK1 was indicated as a target gene of miR-1182 and let-7a. Importantly, upregulation of either miR-1182 or let-7a induced autophagy, and inhibited cell progression and in vivo tumor growth and lung metastasis; moreover, combined treatment of miR-1182 and let-7a overexpression presented with enhanced inhibitory efect on NUAK1 expression and CCA progression, but such synergistic efect could be reversed by overexpression of NUAK1. -

Mutations Suggests Both Loss-Of-Function and Gain-Of-Function Effects Morag A
© 2021. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2021) 14, dmm047225. doi:10.1242/dmm.047225 RESEARCH ARTICLE Hearing impairment due to Mir183/96/182 mutations suggests both loss-of-function and gain-of-function effects Morag A. Lewis1,2,*, Francesca Di Domenico1, Neil J. Ingham1,2, Haydn M. Prosser2 and Karen P. Steel1,2 ABSTRACT birth. However, this is not the cause of the hearing loss; even before The microRNA miR-96 is important for hearing, as point mutations in the onset of normal hearing, homozygote hair cells fail to mature humans and mice result in dominant progressive hearing loss. Mir96 both morphologically and physiologically, remaining in their is expressed in sensory cells along with Mir182 and Mir183, but the immature state, and heterozygote hair cells show a developmental roles of these closely-linked microRNAs are as yet unknown. Here, delay. miR-96 is thus thought to be responsible for coordinating we analyse mice carrying null alleles of Mir182, and of Mir183 and hair cell maturation (Chen et al., 2014; Kuhn et al., 2011). Mir96 together to investigate their roles in hearing. We found that Overexpression of the three miRNAs also results in cochlear defects Mir183/96 heterozygous mice had normal hearing and homozygotes and hearing loss (Weston et al., 2018). The complete loss of all were completely deaf with abnormal hair cell stereocilia bundles and mature miRNAs from the inner ear results in early developmental reduced numbers of inner hair cell synapses at 4 weeks of age. defects including a severely truncated cochlear duct (Friedman Mir182 knockout mice developed normal hearing then exhibited et al., 2009; Soukup et al., 2009). -

Timo Lassmann, Yoshiko Maida 2, Yasuhiro Tomaru, Mami Yasukawa
Int. J. Mol. Sci. 2015, 16, 1192-1208; doi:10.3390/ijms16011192 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article Telomerase Reverse Transcriptase Regulates microRNAs Timo Lassmann 1,†, Yoshiko Maida 2,†, Yasuhiro Tomaru 1, Mami Yasukawa 2, Yoshinari Ando 1, Miki Kojima 1, Vivi Kasim 2, Christophe Simon 1, Carsten O. Daub 1, Piero Carninci 1, Yoshihide Hayashizaki 1,* and Kenkichi Masutomi 2,3,* 1 RIKEN Omics Science Center, RIKEN Yokohama Institute, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan; E-Mails: [email protected] (T.L.); [email protected] (Y.T.); [email protected] (Y.A.); [email protected] (M.K.); [email protected] (C.S.); [email protected] (C.O.D.); [email protected] (P.C.) 2 Cancer Stem Cell Project, National Cancer Center Research Institute, 5-1-1 Tsukiji, Chuo-ku, Tokyo 104-0045, Japan; E-Mails: [email protected] (Y.M.); [email protected] (M.Y.); [email protected] (V.K.) 3 Precursory Research for Embryonic Science and Technology (PREST), Japan Science and Technology Agency, 4-1-8 Honcho Kawaguchi, Saitama 332-0012, Japan † These authors contributed equally to this work. * Authors to whom correspondence should be addressed; E-Mails: [email protected] (Y.H.); [email protected] (K.M.); Tel.: +81-45-503-9248 (Y.H.); +81-3-3547-5173 (K.M.); Fax: +81-45-503-9216 (Y.H.); +81-3-3547-5123 (K.M.). Academic Editor: Martin Pichler Received: 22 November 2014 / Accepted: 26 December 2014 / Published: 6 January 2015 Abstract: MicroRNAs are small non-coding RNAs that inhibit the translation of target mRNAs. -
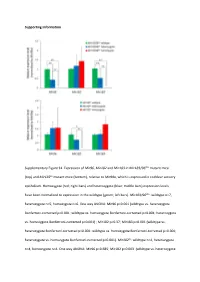
Supporting Information Supplementary Figure S1
Supporting Information Supplementary Figure S1. Expression of Mir96, Mir182 and Mir183 in Mir183/96dko mutant mice (top) and Mir182ko mutant mice (bottom), relative to Mir99a, which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96dko: wildtype n=7, heterozygote n=5, homozygote n=6. One way ANOVA: Mir96 p<0.001 (wildtype vs. heterozygote Bonferroni‐corrected p<0.001; wildtype vs. homozygote Bonferroni‐corrected p<0.001; heterozygote vs. homozygote Bonferroni‐corrected p=0.001) ; Mir182 p=0.37; Mir183 p<0.001 (wildtype vs. heterozygote Bonferroni‐corrected p=0.001; wildtype vs. homozygote Bonferroni‐corrected p<0.001; heterozygote vs. homozygote Bonferroni‐corrected p<0.001). Mir182ko: wildtype n=4, heterozygote n=4, homozygote n=4. One way ANOVA: Mir96 p=0.685; Mir182 p=0.003 (wildtype vs. heterozygote Bonferroni‐corrected p=0.397; wildtype vs. homozygote Bonferroni‐corrected p=0.003; heterozygote vs. homozygote Bonferroni‐corrected p=0.032); Mir183 p=0.04 (wildtype vs. heterozygote Bonferroni‐corrected p=1.0; wildtype vs. homozygote Bonferroni‐corrected p=0.068; heterozygote vs. homozygote Bonferroni‐corrected p=0.094), Error bars are standard deviation (* = P < 0.05, ** = P ≤ 0.01). Supplementary Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. Supplementary Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182ko mice at all ages tested. -

Language Impairment Resulting from a De Novo Deletion of 7Q32.1-Q33: a Case
bioRxiv preprint doi: https://doi.org/10.1101/047241; this version posted April 6, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Language impairment resulting from a de novo deletion of 7q32.1-q33: a case report Mª Salud Jiménez-Romero, Montserrat Barcos-Martínez, Isabel Espejo-Portero, Antonio Benítez-Burraco Corresponding author: Antonio Benítez-Burraco, PhD. Department of Spanish Philology and its Didactics, University of Huelva, Huelva, Spain e-mail: [email protected] Affiliations for the other authors: Mª Salud Jiménez-Romero: Department of Psychology, University of Córdoba, Córdoba, Spain & Maimónides Institute of Biomedical Research, Córdoba, Spain Montserrat Barcos-Martínez: Laboratory of Molecular Genetics, University Hospital “Reina Sofía”, Córdoba, Spain &Maimónides Institute of Biomedical Research, Córdoba, Spain Isabel Espejo-Portero: Laboratory of Molecular Genetics, University Hospital “Reina Sofía”, Córdoba, Spain & Maimónides Institute of Biomedical Research, Córdoba, Spain bioRxiv preprint doi: https://doi.org/10.1101/047241; this version posted April 6, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 2 ABSTRACT Chromosome 7 is a hot spot for cognitive disorders involving language deficits. We report on a girl who presents with a cognitive and speech delay, motor problems, hearing loss, and behavioral disturbances, and a de novo deletion within 7q32.1-q33 (chromosome position: chr7:127109685-132492196, hg 18). Several genes involved in brain development and function are located within the deleted region. -

Noncoding Rnas As Novel Pancreatic Cancer Targets
NONCODING RNAS AS NOVEL PANCREATIC CANCER TARGETS by Amy Makler A Thesis Submitted to the Faculty of The Charles E. Schmidt College of Science In Partial Fulfillment of the Requirements for the Degree of Master of Science Florida Atlantic University Boca Raton, FL August 2018 Copyright 2018 by Amy Makler ii ACKNOWLEDGEMENTS I would first like to thank Dr. Narayanan for his continuous support, constant encouragement, and his gentle, but sometimes critical, guidance throughout the past two years of my master’s education. His faith in my abilities and his belief in my future success ensured I continue down this path of research. Working in Dr. Narayanan’s lab has truly been an unforgettable experience as well as a critical step in my future endeavors. I would also like to extend my gratitude to my committee members, Dr. Binninger and Dr. Jia, for their support and suggestions regarding my thesis. Their recommendations added a fresh perspective that enriched our initial hypothesis. They have been indispensable as members of my committee, and I thank them for their contributions. My parents have been integral to my successes in life and their support throughout my education has been crucial. They taught me to push through difficulties and encouraged me to pursue my interests. Thank you, mom and dad! I would like to thank my boyfriend, Joshua Disatham, for his assistance in ensuring my writing maintained a logical progression and flow as well as his unwavering support. He was my rock when the stress grew unbearable and his encouraging words kept me pushing along. -

Screening for Mutations in Selected Mirna Genes in Hypogonadotropic Hypogonadism Patients
ID: 19-0080 8 5 A-P Iivonen et al. Screening for mutations in 8:5 506–509 miRNA genes RESEARCH Screening for mutations in selected miRNA genes in hypogonadotropic hypogonadism patients Anna-Pauliina Iivonen1, Johanna Känsäkoski1, Kirsi Vaaralahti1 and Taneli Raivio1,2 1Institute of Biomedicine/Physiology, Biomedicum Helsinki and Stem Cells and Metabolism Research Program, University of Helsinki, Helsinki, Finland 2New Children’s Hospital, Pediatric Research Center, Helsinki University Hospital, Helsinki, Finland Correspondence should be addressed to T Raivio: [email protected] Abstract In approximately half of congenital hypogonadotropic hypogonadism (cHH) patients, the Key Words genetic cause remains unidentified. Since the lack of certain miRNAs in animal models f microRNA has led to cHH, we sequenced human miRNAs predicted to regulate cHH-related genes f mutation (MIR7-3, MIR141, MIR429 and MIR200A-C) in 24 cHH patients with Sanger sequencing. A f hypogonadotropic heterozygous variant in MIR200A (rs202051309; general population frequency of 0.02) hypogonadism was found in one patient. Our results suggest that mutations in the studied miRNAs are f Kallmann syndrome unlikely causes of cHH. However, the complex interplay between miRNAs and their target Endocrine Connections genes in these diseases requires further investigations. (2019) 8, 506–509 Introduction Congenital hypogonadotropic hypogonadism (cHH) is a mRNA induces the mRNA’s translational repression rare genetic disease that prevents pubertal development or decay (4). miRNAs from individual gene families and causes infertility due to deficient secretion or action of typically target hundreds of mRNAs, and over 60% of gonadotropin-releasing hormone (GnRH) (1). Congenital human protein-coding genes are miRNA targets (5). -

Micrornas and Inherited Retinal Dystrophies
COMMENTARY COMMENTARY microRNAs and inherited retinal dystrophies Shunbin Xu1 Department of Ophthalmology/Kresge Eye Institute and Department of Anatomy & Cell Biology, School of Medicine, Wayne State University, Detroit, MI 48201 Inherited retinal dystrophies (IRD) are a humans, and the third miRNA in which mu- heterogeneous group of blinding diseases tations cause Mendelian inherited disorders in that affect more than 200,000 Americans humans, after miR-96, which causes nonsyn- and millions worldwide (1). Identification of dromic hearing loss (9, 10), and miR-184 (5, genes that are responsible for IRD when de- 6). Conte et al’s report (8) provides a new fective is of great importance to the basic molecular mechanism of IRD and under- understanding as well as development of ef- scores the importance of miRNA in IRD. ficient gene diagnosis and treatment. Thus miR-204 resides in intron 8 of the tran- Fig. 1. n.37C > T mutation in miR-204 significantly far, 272 genetic loci are linked to various sient receptor potential (TRP) channel gene, changed its predicted target genes. Venn diagram sum- TRPM3 marizing the number of overlapping and unique pre- forms of IRD; among these, 232 protein-cod- , on human chromosome (chr) dicted target genes of wt-miR-204 and mut-miR-204. ing genes have been identified to cause 9q21.12. Through a classic parametric linkage IRD when defective (RetNet, https://sph.uth. analysis, Conte et al. zoomed in on four re- edu/Retnet, last updated April 29, 2015). gions segregating with the phenotypes, in- medaka fish suggested its important roles in microRNAs (miRNAs) are endogenous, cluding 9q21 (8). -

Gain-Of-Function Mutation of Microrna-140 in Human Skeletal Dysplasia
LETTERS https://doi.org/10.1038/s41591-019-0353-2 Gain-of-function mutation of microRNA-140 in human skeletal dysplasia Giedre Grigelioniene1,2,3,15, Hiroshi I. Suzuki 4,15, Fulya Taylan 2, Fatemeh Mirzamohammadi1, Zvi U. Borochowitz5, Ugur M. Ayturk6, Shay Tzur7,8, Eva Horemuzova9,10, Anna Lindstrand2,3, Mary Ann Weis11, Gintautas Grigelionis 2, Anna Hammarsjö 2,3, Elin Marsk12, Ann Nordgren2,3, Magnus Nordenskjöld2,3, David R. Eyre11, Matthew L. Warman 6, Gen Nishimura13, Phillip A. Sharp 4,14 and Tatsuya Kobayashi 1* MicroRNAs (miRNAs) are post-transcriptional regulators of A novel skeletal dysplasia was identified in two unrelated fam- gene expression. Heterozygous loss-of-function point muta- ilies in a project for molecular diagnosis of ultrarare congenital tions of miRNA genes are associated with several human con- skeletal disorders (Fig. 1a,b). In family 1, individual II-2, patient genital disorders1–5, but neomorphic (gain-of-new-function) 1, P1, is affected, as is one of her sons (III-2, P2). In family 2, II-1, mutations in miRNAs due to nucleotide substitutions have not P3 is the only affected individual. Clinical features of the skel- been reported. Here we describe a neomorphic seed region etal dysplasia included disproportionate short stature with short mutation in the chondrocyte-specific, super-enhancer-asso- limbs, small hands and feet, and midface hypoplasia with small ciated MIR140 gene encoding microRNA-140 (miR-140) in nose. The radiological hallmarks were mild spondylar dysplasia, a novel autosomal dominant human skeletal dysplasia. Mice delayed epiphyseal ossification of the hip and knee, and severe with the corresponding single nucleotide substitution show brachydactyly with cone-shaped phalangeal epiphyses (Fig. -

Mir-204 Is Responsible for Inherited Retinal Dystrophy Associated
MiR-204 is responsible for inherited retinal dystrophy PNAS PLUS associated with ocular coloboma Ivan Contea,1, Kristen D. Hadfieldb,1, Sara Barbatoa, Sabrina Carrellaa, Mariateresa Pizzoa, Rajeshwari S. Bhata, Annamaria Carissimoa, Marianthi Karalia, Louise F. Porterb,c, Jill Urquhartb,c, Sofie Hateleyb, James O’Sullivanb, Forbes D. C. Mansonb, Stephan C. F. Neuhaussd, Sandro Banfia,e,f,2, and Graeme C. M. Blackb,c,f,g,2 aTelethon Institute of Genetics and Medicine, Pozzuoli 80078, Italy; bManchester Centre for Genomic Medicine, Institute of Human Development, Faculty of Medical and Human Sciences, University of Manchester, Manchester M13 9WL, United Kingdom; cManchester Centre for Genomic Medicine, St. Mary’s Hospital, Central Manchester University Hospitals National Health Service Foundation Trust, Manchester Academic Health Sciences Centre, Manchester M13 9WL, United Kingdom; dInstitute of Molecular Life Sciences, University of Zurich, CH-8057 Zurich, Switzerland; eMedical Genetics, Department of Biochemistry, Biophysics and General Pathology, Second University of Naples, 80138 Naples, Italy; fEuropean Retinal Dystrophy Consortium; and gUnited Kingdom Inherited Retinal Disease Consortium, Manchester Centre for Genomic Medicine, St. Mary’s Hospital, Central Manchester University Hospitals National Health Service Foundation Trust, Manchester Academic Health Sciences Centre, Manchester M13 9WL, United Kingdom Edited by Jeremy Nathans, Johns Hopkins University, Baltimore, MD, and approved May 11, 2015 (received for review January 29, 2014) Ocular developmental disorders, including the group classified as MicroRNAs (miRNAs) are emerging as key players in the con- microphthalmia, anophthalmia, and coloboma (MAC) and inherited trol of fundamental biological processes in both physiological and retinal dystrophies, collectively represent leading causes of heredi- pathological conditions. They are single-stranded, noncoding, short tary blindness.