Gain-Of-Function Mutation of Microrna-140 in Human Skeletal Dysplasia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Molecular and Genetic Analysis of Otosclerosis
A molecular and genetic analysis of otosclerosis Joanna Lauren Ziff Submitted for the degree of PhD University College London January 2014 1 Declaration I, Joanna Ziff, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Where work has been conducted by other members of our laboratory, this has been indicated by an appropriate reference. 2 Abstract Otosclerosis is a common form of conductive hearing loss. It is characterised by abnormal bone remodelling within the otic capsule, leading to formation of sclerotic lesions of the temporal bone. Encroachment of these lesions on to the footplate of the stapes in the middle ear leads to stapes fixation and subsequent conductive hearing loss. The hereditary nature of otosclerosis has long been recognised due to its recurrence within families, but its genetic aetiology is yet to be characterised. Although many familial linkage studies and candidate gene association studies to investigate the genetic nature of otosclerosis have been performed in recent years, progress in identifying disease causing genes has been slow. This is largely due to the highly heterogeneous nature of this condition. The research presented in this thesis examines the molecular and genetic basis of otosclerosis using two next generation sequencing technologies; RNA-sequencing and Whole Exome Sequencing. RNA–sequencing has provided human stapes transcriptomes for healthy and diseased stapes, and in combination with pathway analysis has helped identify genes and molecular processes dysregulated in otosclerotic tissue. Whole Exome Sequencing has been employed to investigate rare variants that segregate with otosclerosis in affected families, and has been followed by a variant filtering strategy, which has prioritised genes found to be dysregulated during RNA-sequencing. -

Whole Genome Sequencing Identifies Novel Structural Variant in a Large Indian Family Affected with X - Linked Agammaglobulinemia
medRxiv preprint doi: https://doi.org/10.1101/2020.10.05.20200949; this version posted October 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-ND 4.0 International license . Whole Genome Sequencing identifies novel structural variant in a large Indian family affected with X - linked agammaglobulinemia 1,2,# 3,5,#,$ 3,4 3 Abhinav Jain , Geeta Madathil Govindaraj , Athulya Edavazhippurath , Nabeel Faisal , Rahul C 1 1,2 6 3 1 1 Bhoyar , Vishu Gupta , Ramya Uppuluri , Shiny Padinjare Manakkad , Atul Kashyap , Anoop Kumar , 1,2 1,2 7 7 3 Mohit Kumar Divakar , Mohamed Imran , Sneha Sawant , Aparna Dalvi , Krishnan Chakyar , 7 6 1,2,$ 1,2,$ Manisha Madkaikar , Revathi Raj , Sridhar Sivasubbu , Vinod Scaria 1 CSIR-Institute of Genomics and Integrative Biology, Mathura Road, New Delhi, Delhi, India 2 Academy of Scientific and Innovative Research (AcSIR), Mathura Road, New Delhi, Delhi, India 3 Department of Pediatrics, Government Medical College Kozhikode, Kozhikode, Kerala, India 4 Multidisciplinary Research Unit, Government College Kozhikode, Kozhikode, Kerala, India 5 FPID Regional Diagnostic Centre, Government Medical College Kozhikode, Kozhikode, Kerala India 6 Department of Pediatric Hematology, Oncology, Blood and Marrow Transplantation, Apollo Hospitals, 320, Padma complex, Anna salai, Teynampet, Chennai, Tamil Nadu, India 7 Department of Pediatric Immunology and Leukocyte Biology, ICMR-National Institute of Immunohaematology, KEM Hospital, Parel, Mumbai, Maharashtra, India. # Joint first authors $ Corresponding author Vinod Scaria: [email protected] Sridhar Sivasubbu: [email protected] Geeta Madathil Govindaraj: [email protected] NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. -

Mir-96 Regulates the Progression of Differentiation in Mammalian Cochlear Inner and Outer Hair Cells
miR-96 regulates the progression of differentiation in mammalian cochlear inner and outer hair cells Stephanie Kuhna,1, Stuart L. Johnsona,1, David N. Furnessb, Jing Chenc, Neil Inghamc, Jennifer M. Hiltonc, Georg Steffesc, Morag A. Lewisc, Valeria Zampinia,d, Carole M. Hackneya, Sergio Masettod, Matthew C. Holleya, Karen P. Steelc, and Walter Marcottia,2 aDepartment of Biomedical Science, University of Sheffield, Sheffield S10 2TN, United Kingdom; bInstitute for Science and Technology in Medicine, Keele University, Keele ST5 5BG, United Kingdom; cWellcome Trust Sanger Institute, Hinxton, Cambridge CB10 1SA, United Kingdom; and dDepartment of Physiology, University of Pavia, 27100 Pavia, Italy Edited by Mary-Claire King, University of Washington, Seattle, WA, and approved December 27, 2010 (received for review November 8, 2010) MicroRNAs (miRNAs) are small noncoding RNAs able to regulate cells up to at least P5 (23) and in the spiral ganglion up to P14 a broad range of protein-coding genes involved in many biological (19). Mutations in miR-96 have been associated with non- processes. miR-96 is a sensory organ-specific miRNA expressed in syndromic progressive hearing loss in humans (15) and mice (23). the mammalian cochlea during development. Mutations in miR-96 We found that hair cell development in diminuendo mice with cause nonsyndromic progressive hearing loss in humans and mice. a Mir96 mutation is arrested at around the day of birth, a time The mouse mutant diminuendo has a single base change in the when IHCs and OHCs still exhibit qualitatively similar bio- seed region of the Mir96 gene leading to widespread changes in physical properties. -

Microrna-1182 and Let-7A Exert Synergistic Inhibition on Invasion
Pan et al. Cancer Cell Int (2021) 21:161 https://doi.org/10.1186/s12935-021-01797-z Cancer Cell International PRIMARY RESEARCH Open Access MicroRNA-1182 and let-7a exert synergistic inhibition on invasion, migration and autophagy of cholangiocarcinoma cells through down-regulation of NUAK1 Xin Pan*, Gang Wang and Baoming Wang Abstract Background: Cholangiocarcinoma (CCA) is the second most common primary liver malignancy worldwide. Several microRNAs (miRNAs) have been implicated as potential tumor suppressors in CCA. This study aims to explore the potential efects of miR-1182 and let-7a on CCA development. Methods: Bioinformatics analysis was conducted to screen diferentially expressed genes in CCA, Western blot analysis detected NUAK1 protein expression and RT-qPCR detected miR-1182, let-7a and NUAK1 expression in CCA tissues and cell lines. Dual luciferase reporter gene assay and RIP were applied to validate the relationship between miR-1182 and NUAK1 as well as between let-7a and NUAK1. Functional experiment was conducted to investigate the role of miR-1182, let-7a and NUAK1 in cell migration, proliferation and autophagy. Then, the CCA cells that received various treatments were implanted to mice to establish animal model, followed by tumor observation and HE staining to evaluate lung metastasis. Results: CCA tissues and cells were observed to have a high expression of NUAK1 and poor expression of miR-1182 and let-7a. NUAK1 was indicated as a target gene of miR-1182 and let-7a. Importantly, upregulation of either miR-1182 or let-7a induced autophagy, and inhibited cell progression and in vivo tumor growth and lung metastasis; moreover, combined treatment of miR-1182 and let-7a overexpression presented with enhanced inhibitory efect on NUAK1 expression and CCA progression, but such synergistic efect could be reversed by overexpression of NUAK1. -

Exome Sequencing and Functional Validation in Zebrafish Identify
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REPORT Exome Sequencing and Functional Validation in Zebrafish Identify GTDC2 Mutations as a Cause of Walker-Warburg Syndrome M. Chiara Manzini,1,2 Dimira E. Tambunan,1,2 R. Sean Hill,1,2 Tim W. Yu,1,2 Thomas M. Maynard,3 Erin L. Heinzen,4 Kevin V. Shianna,4 Christine R. Stevens,5 Jennifer N. Partlow,1,2 Brenda J. Barry,1,2 Jacqueline Rodriguez,1,2 Vandana A. Gupta,1,6 Abdel-Karim Al-Qudah,7 Wafaa M. Eyaid,8 Jan M. Friedman,9,10 Mustafa A. Salih,11 Robin Clark,12 Isabella Moroni,13 Marina Mora,14 Alan H. Beggs,1,6 Stacey B. Gabriel,5 and Christopher A. Walsh1,2,5,* Whole-exome sequencing (WES), which analyzes the coding sequence of most annotated genes in the human genome, is an ideal approach to studying fully penetrant autosomal-recessive diseases, and it has been very powerful in identifying disease-causing muta- tions even when enrollment of affected individuals is limited by reduced survival. In this study, we combined WES with homozygosity analysis of consanguineous pedigrees, which are informative even when a single affected individual is available, to identify genetic mutations responsible for Walker-Warburg syndrome (WWS), a genetically heterogeneous autosomal-recessive disorder that severely affects the development of the brain, eyes, and muscle. Mutations in seven genes are known to cause WWS and explain 50%–60% of cases, but multiple additional genes are expected to be mutated because unexplained cases show suggestive linkage to diverse loci. -

Genomic Approaches for Understanding the Genetics of Complex Disease
Downloaded from genome.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Perspective Genomic approaches for understanding the genetics of complex disease William L. Lowe Jr.1 and Timothy E. Reddy2,3 1Division of Endocrinology, Metabolism and Molecular Medicine, Department of Medicine, Northwestern University Feinberg School of Medicine, Chicago, Illinois 60611, USA; 2Department of Biostatistics and Bioinformatics, Duke University Medical School, Durham, North Carolina 27708, USA; 3Center for Genomic and Computational Biology, Duke University Medical School, Durham, North Carolina 27708, USA There are thousands of known associations between genetic variants and complex human phenotypes, and the rate of novel discoveries is rapidly increasing. Translating those associations into knowledge of disease mechanisms remains a fundamental challenge because the associated variants are overwhelmingly in noncoding regions of the genome where we have few guiding principles to predict their function. Intersecting the compendium of identified genetic associations with maps of regulatory activity across the human genome has revealed that phenotype-associated variants are highly enriched in candidate regula- tory elements. Allele-specific analyses of gene regulation can further prioritize variants that likely have a functional effect on disease mechanisms; and emerging high-throughput assays to quantify the activity of candidate regulatory elements are a promising next step in that direction. Together, these technologies have created the ability to systematically and empirically test hypotheses about the function of noncoding variants and haplotypes at the scale needed for comprehensive and system- atic follow-up of genetic association studies. Major coordinated efforts to quantify regulatory mechanisms across genetically diverse populations in increasingly realistic cell models would be highly beneficial to realize that potential. -

QTL Analysis Reveals Genomic Variants Linked to High-Temperature
Wang et al. Biotechnol Biofuels (2019) 12:59 https://doi.org/10.1186/s13068-019-1398-7 Biotechnology for Biofuels RESEARCH Open Access QTL analysis reveals genomic variants linked to high-temperature fermentation performance in the industrial yeast Zhen Wang1,2†, Qi Qi1,2†, Yuping Lin1*, Yufeng Guo1, Yanfang Liu1,2 and Qinhong Wang1* Abstract Background: High-temperature fermentation is desirable for the industrial production of ethanol, which requires thermotolerant yeast strains. However, yeast thermotolerance is a complicated quantitative trait. The understanding of genetic basis behind high-temperature fermentation performance is still limited. Quantitative trait locus (QTL) map- ping by pooled-segregant whole genome sequencing has been proved to be a powerful and reliable approach to identify the loci, genes and single nucleotide polymorphism (SNP) variants linked to quantitative traits of yeast. Results: One superior thermotolerant industrial strain and one inferior thermosensitive natural strain with distinct high-temperature fermentation performances were screened from 124 Saccharomyces cerevisiae strains as parent strains for crossing and segregant isolation. Based on QTL mapping by pooled-segregant whole genome sequencing as well as the subsequent reciprocal hemizygosity analysis (RHA) and allele replacement analysis, we identifed and validated total eight causative genes in four QTLs that linked to high-temperature fermentation of yeast. Interestingly, loss of heterozygosity in fve of the eight causative genes including RXT2, ECM24, CSC1, IRA2 and AVO1 exhibited posi- tive efects on high-temperature fermentation. Principal component analysis (PCA) of high-temperature fermentation data from all the RHA and allele replacement strains of those eight genes distinguished three superior parent alleles including VPS34, VID24 and DAP1 to be greatly benefcial to high-temperature fermentation in contrast to their inferior parent alleles. -

Mutations Suggests Both Loss-Of-Function and Gain-Of-Function Effects Morag A
© 2021. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2021) 14, dmm047225. doi:10.1242/dmm.047225 RESEARCH ARTICLE Hearing impairment due to Mir183/96/182 mutations suggests both loss-of-function and gain-of-function effects Morag A. Lewis1,2,*, Francesca Di Domenico1, Neil J. Ingham1,2, Haydn M. Prosser2 and Karen P. Steel1,2 ABSTRACT birth. However, this is not the cause of the hearing loss; even before The microRNA miR-96 is important for hearing, as point mutations in the onset of normal hearing, homozygote hair cells fail to mature humans and mice result in dominant progressive hearing loss. Mir96 both morphologically and physiologically, remaining in their is expressed in sensory cells along with Mir182 and Mir183, but the immature state, and heterozygote hair cells show a developmental roles of these closely-linked microRNAs are as yet unknown. Here, delay. miR-96 is thus thought to be responsible for coordinating we analyse mice carrying null alleles of Mir182, and of Mir183 and hair cell maturation (Chen et al., 2014; Kuhn et al., 2011). Mir96 together to investigate their roles in hearing. We found that Overexpression of the three miRNAs also results in cochlear defects Mir183/96 heterozygous mice had normal hearing and homozygotes and hearing loss (Weston et al., 2018). The complete loss of all were completely deaf with abnormal hair cell stereocilia bundles and mature miRNAs from the inner ear results in early developmental reduced numbers of inner hair cell synapses at 4 weeks of age. defects including a severely truncated cochlear duct (Friedman Mir182 knockout mice developed normal hearing then exhibited et al., 2009; Soukup et al., 2009). -

Timo Lassmann, Yoshiko Maida 2, Yasuhiro Tomaru, Mami Yasukawa
Int. J. Mol. Sci. 2015, 16, 1192-1208; doi:10.3390/ijms16011192 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article Telomerase Reverse Transcriptase Regulates microRNAs Timo Lassmann 1,†, Yoshiko Maida 2,†, Yasuhiro Tomaru 1, Mami Yasukawa 2, Yoshinari Ando 1, Miki Kojima 1, Vivi Kasim 2, Christophe Simon 1, Carsten O. Daub 1, Piero Carninci 1, Yoshihide Hayashizaki 1,* and Kenkichi Masutomi 2,3,* 1 RIKEN Omics Science Center, RIKEN Yokohama Institute, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan; E-Mails: [email protected] (T.L.); [email protected] (Y.T.); [email protected] (Y.A.); [email protected] (M.K.); [email protected] (C.S.); [email protected] (C.O.D.); [email protected] (P.C.) 2 Cancer Stem Cell Project, National Cancer Center Research Institute, 5-1-1 Tsukiji, Chuo-ku, Tokyo 104-0045, Japan; E-Mails: [email protected] (Y.M.); [email protected] (M.Y.); [email protected] (V.K.) 3 Precursory Research for Embryonic Science and Technology (PREST), Japan Science and Technology Agency, 4-1-8 Honcho Kawaguchi, Saitama 332-0012, Japan † These authors contributed equally to this work. * Authors to whom correspondence should be addressed; E-Mails: [email protected] (Y.H.); [email protected] (K.M.); Tel.: +81-45-503-9248 (Y.H.); +81-3-3547-5173 (K.M.); Fax: +81-45-503-9216 (Y.H.); +81-3-3547-5123 (K.M.). Academic Editor: Martin Pichler Received: 22 November 2014 / Accepted: 26 December 2014 / Published: 6 January 2015 Abstract: MicroRNAs are small non-coding RNAs that inhibit the translation of target mRNAs. -
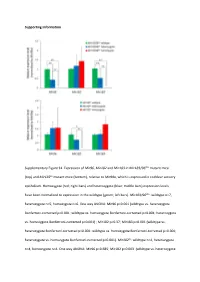
Supporting Information Supplementary Figure S1
Supporting Information Supplementary Figure S1. Expression of Mir96, Mir182 and Mir183 in Mir183/96dko mutant mice (top) and Mir182ko mutant mice (bottom), relative to Mir99a, which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96dko: wildtype n=7, heterozygote n=5, homozygote n=6. One way ANOVA: Mir96 p<0.001 (wildtype vs. heterozygote Bonferroni‐corrected p<0.001; wildtype vs. homozygote Bonferroni‐corrected p<0.001; heterozygote vs. homozygote Bonferroni‐corrected p=0.001) ; Mir182 p=0.37; Mir183 p<0.001 (wildtype vs. heterozygote Bonferroni‐corrected p=0.001; wildtype vs. homozygote Bonferroni‐corrected p<0.001; heterozygote vs. homozygote Bonferroni‐corrected p<0.001). Mir182ko: wildtype n=4, heterozygote n=4, homozygote n=4. One way ANOVA: Mir96 p=0.685; Mir182 p=0.003 (wildtype vs. heterozygote Bonferroni‐corrected p=0.397; wildtype vs. homozygote Bonferroni‐corrected p=0.003; heterozygote vs. homozygote Bonferroni‐corrected p=0.032); Mir183 p=0.04 (wildtype vs. heterozygote Bonferroni‐corrected p=1.0; wildtype vs. homozygote Bonferroni‐corrected p=0.068; heterozygote vs. homozygote Bonferroni‐corrected p=0.094), Error bars are standard deviation (* = P < 0.05, ** = P ≤ 0.01). Supplementary Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. Supplementary Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182ko mice at all ages tested. -

A Comprehensive Database of Putative Human Microrna Target Site
bioRxiv preprint doi: https://doi.org/10.1101/554485; this version posted May 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. dbMTS: a comprehensive database of putative human microRNA target site SNVs and their functional predictions Chang Li1,2, Michael D. Swartz3, Bing Yu1, Yongsheng Bai4, Xiaoming Liu1,2* 1Human Genetics Center and Department of Epidemiology, Human Genetics and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 2USF Genomics, College of Public Health, University of South Florida, Tampa, FL 3Department of Biostatistics, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 4Department of Internal Medicine, University of Michigan, Ann Arbor, MI *Correspondence: Xiaoming Liu, [email protected] Funding information: This work is supported partially by NIH funding 1UM1HG008898 and partially by the startup funding for XL from the University of South Florida. bioRxiv preprint doi: https://doi.org/10.1101/554485; this version posted May 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract microRNAs (miRNAs) are short non-coding RNAs that can repress the expression of protein coding messenger RNAs (mRNAs) by binding to the 3’UTR of the target. -

The Role of Natural Selection in Human Evolution – Insights from Latin America
Genetics and Molecular Biology, 39, 3, 302-311 (2016) Copyright © 2016, Sociedade Brasileira de Genética. Printed in Brazil DOI: http://dx.doi.org/10.1590/1678-4685-GMB-2016-0020 Review Article The role of natural selection in human evolution – insights from Latin America Francisco M. Salzano1 1Departamento de Genética, Instituto de Biociências, Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre, RS, Brazil. Abstract A brief introduction considering Darwin’s work, the evolutionary synthesis, and the scientific biological field around the 1970s and subsequently, with the molecular revolution, was followed by selected examples of recent investiga- tions dealing with the selection-drift controversy. The studies surveyed included the comparison between essential genes in humans and mice, selection in Africa and Europe, and the possible reasons why females in humans remain healthy and productive after menopause, in contrast with what happens in the great apes. At the end, selected exam- ples of investigations performed in Latin America, related to the action of selection for muscle performance, acetylation of xenobiotics, high altitude and tropical forest adaptations were considered. Despite dissenting views, the influence of positive selection in a considerable portion of the human genome cannot presently be dismissed. Keywords: natural selection, human evolution, population genetics, human adaptation, history of genetics. Received: February 10, 2016; Accepted: May 8, 2016. History deniably indicated that we had derived from