Extraction of Polar Pollutants Using
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Solvent Extraction Instruments Brochure
ROT-X-TRACT SERİES Solvent Extraction Instruments PRODUCT BROCHURE ELEMENTEL ANALİTİK ve BİO TEKNOLOJİK SİSTEMLER | Folkart Towers - Adalet Mah. Manas Blv. No: 39/3408 Bayraklı/IZMIR, TURKEY, PHONE-FAX: +90 232 472 17 11 • [email protected] • www.elementel.com 2 Content www.elementel.com CONTENT ORGANOMATION SOLVENT EXTRACTORS OVERVIEW ..................................................................................... 3 ROT-X-TRACT-S SERIES ROTARY SOLID-LIQUID SOXHLET EXTRACTORS ........................................................ 5 ROT-X-TRACT-LC SERIES CORNING ACCELERATED ONE-STEP LIQUID-LIQUID EXTRACTORS ....................... 7 ROT-X-TRACT SOLVENT EXTRACTION INSTRUMENTS ACCESSORIES AND REPLACEMENT PARTS ............ 9 Organomation Solvent Extraction Instruments Overview 3 www.elementel.com Organomation Solvent Extractors Overview You are in the lab and need to run multiple extractions at once. First you set the Soxhlet extractors up on individual heating mantles or baths. Next, tubing is individually attached to each of the condensers. There are dozens of tubes to deal with, and multiple heating mantles to monitor. The problem with this arrangement is that in the end, you have spent more time assembling and monitoring your apparatuses than performing the actual chemistry. To conserve valuable bench space, all samples are arranged in a circle and the instrument rotates allowing each sample to be accessed from the front. Individual condensers are connected to the centrally located water manifold through quick disconnect fittings. -

Method 23 Determination of Polychlorinated Dibenzo-P-Dioxins
Method 23 8/3/2017 While we have taken steps to ensure the accuracy of this Internet version of the document, it is not the official version. To see a complete version including any recent edits, visit: https://www.ecfr.gov/cgi-bin/ECFR?page=browse and search under Title 40, Protection of Environment. METHOD 23—DETERMINATION OF POLYCHLORINATED DIBENZO-P-DIOXINS AND POLYCHLORINATED DIBENZOFURANS FROM STATIONARY SOURCES 1. Applicability and Principle 1.1 Applicability. This method is applicable to the determination of polychlorinated dibenzo-p- dioxins (PCDD's) and polychlorinated dibenzofurans (PCDF's) from stationary sources. 1.2 Principle. A sample is withdrawn from the gas stream isokinetically and collected in the sample probe, on a glass fiber filter, and on a packed column of adsorbent material. The sample cannot be separated into a particle vapor fraction. The PCDD's and PCDF's are extracted from the sample, separated by high resolution gas chromatography, and measured by high resolution mass spectrometry. 2. Apparatus 2.1 Sampling. A schematic of the sampling train used in this method is shown in Figure 23-1. Sealing greases may not be used in assembling the train. The train is identical to that described in section 2.1 of Method 5 of this appendix with the following additions: Method 23 8/3/2017 2.1.1 Nozzle. The nozzle shall be made of nickel, nickel-plated stainless steel, quartz, or borosilicate glass. 2.1.2 Sample Transfer Lines. The sample transfer lines, if needed, shall be heat traced, heavy 1 1 walled TFE ( ⁄2 in. -

Soxhlet Extraction of Solid Materials: an Outdated Technique with a Promising Innovative Future
Analytica Chimica Acta 369 (1998) 1±10 Soxhlet extraction of solid materials: an outdated technique with a promising innovative future M.D. Luque de Castro*, L.E. GarcõÂa-Ayuso Department of Analytical Chemistry, Faculty of Sciences, University of CoÂrdoba, E-14004 CoÂrdoba, Spain Received 8 December 1997; received in revised form 23 March 1998; accepted 30 March 1998 Abstract An overview of the evolution of Soxhlet extraction of solid materials and its comparison with the performance of other conventional and new extraction techniques is presented. First, a discussion on both conventional Soxhlet as compared with other conventional extraction techniques and some minor improvements of the former for speci®c applications is done. Secondly, a critical comparison of conventional Soxhlet with the new extraction techniques such as supercritical ¯uid extraction, microwave-assisted processes and microwave-assisted solvent extraction shows the reasons why major, recent improvements of this technique (namely Soxtec1 System HT, Soxwave-100 and focused microwave-assisted Soxhlet extraction) have been proposed, aimed at overcoming most of the shortcomings of conventional Soxhlet and converting it into an updated tool for leaching which competes advantageously with the most recent alternatives in the extraction ®eld. # 1998 Elsevier Science B.V. All rights reserved. Keywords: Updated Soxhlet; Review 1. Introduction physicochemical terminology, as leaching or lixivia- tion, is one of the oldest ways of solid sample pre- Sample pretreatment is often one of the most time treatment. Among the techniques used for consuming steps of the analytical process, particularly implementation of this step, Soxhlet has been the when solid samples are involved. The search for leaching technique mostly used for a long time. -

Hyoscyami Semen (Unprocessed)(Unprocessed)
HyoscyamiHyoscyami Semen (unprocessed)(unprocessed) B CC 0.50.5 mm mm AA 11 mm FigureFigure 1 1 A A photographphotograph of Hyoscyami Semen (unprocessed)(unprocessed) A.A. Hyoscyami Hyoscyami SemenSemen (unprocessed)(unprocessed) B. Magnified image of seeds B. Magnified image of seeds C. Magnified image of longitudinal section of seeds C. Magnified image of longitudinal section of seeds 175 175 Hyoscyami Semen (unprocessed) Hyoscyami Semen (unprocessed) 1. NAMES Powder Exotesta cells scattered or in groups, yellow or greyish-yellowish brown, polygonal or elongated Official Name: Hyoscyami Semen (unprocessed) polygonal in surface view, 75-311 µm long, 25-123 µm in diameter, anticlinal walls thickened, Chinese Name: undulant, with distinct striations, containing yellowish-brown contents; bright yellowish-white under the polarized microscope. Endosperm cells polygonal or subrounded in surface view, cell Chinese Phonetic Name: Tianxianzi (Sheng) walls slightly thickened, containing aleurone grains and oil droplets. Cotyledon cells colourless, subpolygonal, with thin walls, containing oil droplets (Fig. 3). 2. SOURCE Hyoscyami Semen (unprocessed) is the unprocessed dried ripe seed of Hyoscyamus niger L. (Solanaceae). The fruit is collected in summer and autumn when the pericarp turns yellow, exposed to the sun, afterwards the seeds tapped out, pericarp and stalk removed, then dried under the sun to obtain Hyoscyami Semen (unprocessed). 3. DESCRIPTION Flattened-subreniform to flattened-ovoid, about 1 mm in diameter. Externally brownish-yellow or greyish-yellow, with fine and dense reticulate striations, and a pitted hilum located at the slightly acute end. Cut surface greyish-white, oily, containing endosperm, embryo curved. Odour slight (Fig. 1). 4. IDENTIFICATION 4.1 Microscopic Identification (Appendix III) Transverse section Exotesta cells with irregularly undulant protuberances, apex of the protuberances tapering or blunt, cell walls with transparent striations. -

Seed Oil Using Soxhlet Method
S.H. Mohd-SetaparMalaysian et al. Journal / Malaysian of Fundamental Journal of andFundamental Applied Sciences and Applied Vol.10, Sciences No.1 (2014)Vol.10, 1-6 No.1 (2014) 1-6 Extraction of rubber (Hevea brasiliensis) seed oil using soxhlet method S.H. Mohd-Setapar*, Lee Nian-Yian and N.S. Mohd-Sharif Centre of Lipid Engineering & Applied Research (CLEAR), Dept. of Chemical Eng., Fac. of Chemical Eng., Universiti Teknologi Malaysia 81310 Skudai, Johor, Malaysia. *Corresponding Author: [email protected] (S.H. Mohd-Setapar) Article history : ABSTRACT Received 4 January 2013 Revised 24 June 2013 Soxhlet extraction which is also known as solvent extraction refers to the preferential dissolution of oil by Accepted 1 July 2013 contacting oilseeds with a liquid solvent. This is the most efficient method to recover oil from oilseeds, Available online 1 August 2013 thus solvent extraction using hexane has been commercialized as a standard practice in today’s industry. In this study, soxhlet extraction had been used to extract the rubber seed oil which contains high GRAPHICAL ABSTRACT percentage of alpha-linolenic acid. In addition, the different solvents will be used for the extraction of rubber seed oil such as petroleum ether, n-hexane, ethanol and water to study the best solvent to extract the rubber seed oil so the maximum oil yield can be obtained. On the other hands, the natural resource, rubber belongs to the family of Euphorbiaceae, the genus is Hevea while the species of rubber is brasiliensis. Rubber (Hevea brasiliensis) seeds are abundant and wasted because they had not been used in any industry or applications in daily life. -

Soxhlet Extraction Classic Fat Determination with Soxhlet Extraction After Hydrolysis
Soxhlet extraction Classic fat determination with Soxhlet extraction after hydrolysis Hydrolysis principle This acid digestion process dissolves both "free fats" as well as "bound fats" from the overall fat content. The fat is frequently naturally enclosed in the cell matrix of the food or fodder or chemically bound. In these cases, a hydrolysis step before extraction completely releases the fat. Hydrolysis-digestion units p. 18 The user filters the hydrolysate of the separated sample by using a glass sample tube filled with sand or Celite. The user then rinses the fatty filter residue with water in order to remove the acid. Filter equipment for hydrolysis p. 18 After drying, the filter residue is finally extracted. Soxhlet extraction systems p. 9 8 Soxhlet Complete single extraction units The standard extraction method is the Soxhlet method. behr apparatus for Soxhlet extractions fulfil all the various requirements in everyday laboratory practice. n Practical brackets for condensers and intermediate extraction pieces for safe storage between extractions n Extractor sizes from 30 ml to 1,000 ml n Compact apparatus with one sample position n Series extraction devices with 4, 6 or 8 sample positions n Extractors with specially developed siphon tubes (make: "Bröckerhoff") guarantee consistent extraction cycles across all sample positions n Extractors with taps remove the need for additional distillation after the extraction n Condensers with threaded fittings The behr hydrolysis units (4 or 6 sample positions) also enable acid digestion prior to extraction (determination of the total fat content according to Weibull and Stoldt). Complete single extraction units Complete single extraction units with base frame, heating device, bracket, tubes and glass apparatus (reaction flask, extractor, Dimroth condenser for extraction). -

Procedure for Analysis * of Nickel-Cadmium Cell Materials
Z r - X-761-73-314 PREPRINT PROCEDURE FOR ANALYSIS * OF NICKEL-CADMIUM CELL MATERIALS ( (NASA-TM-X-70575) PROCEDURE FOR ANALYSIS N74-15744 OF NICKEL-CADMIUM CELL MATEBIALS (NASA) 46 p HC $4.50 CSCL 10C Unclas - 3/ 28984 GERALD HALPERT OLAMIDE OGUNYANKIN CLENTON JONES OCTOBER 1973, Jeproduced by NATIONAL TECHNICAL ___ INFORMATION SERVICE -: "" Springfield, V A . 2215 GODDARD SPACE FLIGHT CENTER 'GREENBELT , MARYLAND 'C'-t - - I - - X-761-73-314 PROCEDURE FOR ANALYSIS OF NICKEL-CADMIUM CELL MATERIALS Gerald Halpert Goddard Space Flight Center Olamide Ogunyankin and Clenton Jones Federal City College October 1973 GODDARD SPACE FLIGHT CENTER Greenbelt, Maryland / CONTENTS Preceding page blank Page INTRODUCTION ...................... 1 1. MANUFACTURING DATA, CELL HISTORY . 3 2. VISUAL/MECHANICAL INSPECTION OF CELL . 3 3. GAS SAMPLING AND ANALYSIS . .......... 4 4. CELL DISASSEMBLY, SEPARATOR SAMPLE REMOVAL, SOXHLET EXTRACTION, AND PLATE DRYING . 4 4.1 Equipment .. .................... 4 4.2 Procedure . .. .................. 5 5. ANALYSIS OF SEPARATOR SAMPLES FOR OH AND CO 3 . 6 5.1 Principle . 6 5.2 Reagents . .. ..... .. 7 5.3 Procedure . ... 7 5.4 Calculations . 8 6. ELECTROLYTE EXTRACT ANALYSIS . 8 6.1 Principle . 8 6.2 Reagents . 9 6.3 Procedure . ... 9 6.4 Calculations . 9 7. PLATE IDENTIFICATION, WEIGHT, AND THICKNESS . 9 8. ANALYSIS OF THE NEGATIVE PLATE . 10 8.1 Principle . 10 8.2 Reagents . ...... 10 8.3 Procedure ...................... 12 8.4 Analysis for Cadmium Metal - Charged State . 14 8.5 Calculations .. .................. 15 iii CONTENTS (continued) Page 9. CHEMICAL ANALYSIS OF POSITIVE PLATES . ....... 16 9.1 Principle ......................... 16 9.2 Reagents . .. 17 9. 3 Charged Material Analysis . ...... 17 9.4 Total Active Material and Metallic Nickel Analysis . -

Comparative Analysis on the Extraction of Essential Oil from Lemongrass and Basil Leaves
IJISET - International Journal of Innovative Science, Engineering & Technology, Vol. 5 Issue 11, November 2018 ISSN (Online) 2348 – 7968 www.ijiset.com Comparative Analysis on the Extraction of Essential Oil from Lemongrass and Basil Leaves Abbas Mustapha Department of Petroleum and Natural Gas Processing, Petroleum Training Institute, Effurun, Nigeria Abstract Essential oils are generally of botanical extracts of This present work is focused on the extraction of essential oil various plant materials, and do not only originated from from lemongrass and basil leave. Lemongrass and basil leaves flowers, but from herbs, trees and various other plant were obtained from Ugbomoro community, Uwie Local materials. It is also known as the volatile, odoriferous oil Government Delta State, Nigeria. The samples were air dried at of vegetable origin or simply as the “oil of’ the plant from room temperature for about three weeks to reduce the moisture content. The dried leave sample were pressurized (blended) and which they were extracted [6]. From estimate gotten from then used for the extraction processes. 500g of the samples global number of plants; plants are of the order of 300,000 yielded 2.5% and 1.89% respectively by soxhiet extraction and about 10% of these contains essential oils and could be method. The extracted oil were characterized for acid value used as a source for their production [7]. Their extracts are (Lemongrass oil: 4.09 MgKOHJg, Basil leaves oil : 3.95 formed by combination of diverse and complex volatile MgJCOHIg), boiling point (Lemongrass oil 299°C, Basil leaves mixtures of chemical compounds mainly of terpene oil: 215°C), saponicatioin value (Lemongrass oil: 143 associated to aldehydes, alcohols, and ketones which were MgKOHJg , Basil oil: 19SMgKOHIg), peroxide value (Lemon accumulated in various structure of the plant [8]. -

Review of Extraction Techniques
International Journal of Advanced Research in Chemical Science (IJARCS) Volume 6, Issue 3, 2019, PP 6-21 ISSN No. (Online) 2349-0403 DOI: http://dx.doi.org/10.20431/2349-0403.0603002 www.arcjournals.org Review of Extraction Techniques Extraction Methods: Microwave, Ultrasonic, Pressurized Fluid, Soxhlet Extraction, Etc Komal Patel1, Namrata Panchal2, Dr. Pradnya Ingle3* 3Associate Professor Department of Chemical Engineering, Shivajirao S. Jondhale College of Engineering, Dombivli (East), 421201, University of Mumbai, India. *Corresponding Author: Dr. Pradnya Ingle, Associate Professor Department of Chemical Engineering, Shivajirao S. Jondhale College of Engineering, Dombivli (East), 421201, University of Mumbai, India. Abstract: In recent years, variety of Extraction techniques has been introduced for the recovery of organic compounds. Extraction Methods are widely used in various Industries for Separation of components and has wide range of applications. Details of basic theories applicable to types of Extraction such as - Liquid- Liquid Extraction, Solid Phase Extraction, Solid Liquid Extraction and Supercritical Extraction, etc. including the choice of solvent, procedure, respective advantages disadvantages and their applications are explained. Finally, the specific extraction techniques such as Microwave Extraction, Ultrasonic Extraction, Pressurized Fluid Extraction and Soxhlet Extraction along with their applications are also explained. Keywords: Extraction, Liquid-Liquid Extraction, Solid Phase Extraction, Solid Liquid Extraction, -
0013 Extraction App 98-99 New FINAL 23 06 2020.Cdr
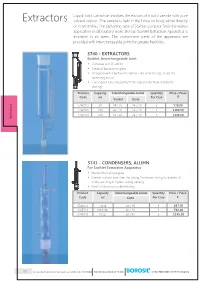
Liquid Solid Extraction involves the elution of a solid sample with pure Extractors solvent vapour. The sample is held in the Extractor body either directly or in a thimble. The siphoning type of Soxhlet Extractor finds the widest application in laboratory work. Borosil Soxhlet Extraction Apparatus is available in six sizes. The component parts of the apparatus are provided with interchangeable joints for greater flexibility. 3740 - EXTRACTORS BOROSIL R Soxhlet, Interchangeable Joint Ÿ Complies with IS 10640 Ÿ Made of borosilicate glass Ÿ Designed with a bulb in the siphon tube near the top, to aid the siphoning action Ÿ The vapour tube also protects the siphon tube from accidental damage Product Capacity Interchangeable Joints Quantity Price / Piece Code ml Per Case ` Socket Cone s 3740013 60 34 / 35 24 / 29 5 778.00 or 3740016 100 40 / 38 24 / 29 5 1100.00 3740020 200 50 / 42 24 / 29 5 1328.00 Extract 3741 - CONDENSERS, ALLIHN For Soxhlet Extraction Apparatus BOROSIL R Ÿ Made of borosilicate glass Ÿ Greater surface area than the Liebig Condenser owing to a series of bulbs, resulting in higher cooling capacity Ÿ Ideal for laboratory scale refluxing Product Capacity Interchangeable Joints Quantity Price / Piece Code ml Cone Per Case ` 3741013 Small 34 / 35 5 637.00 3741016 Medium 40 / 38 5 794.00 3741020 Large 50 / 42 5 1145.00 98 an ISO 9001:2015 certified company For products listed on this page use HSN code 70179090 PRICES EXCLUSIVE OF TAXES 19 3840 - EXTRACTION APPARATUS Soxhlet Complete With Allihn Condenser Interchangeable Joint -

Extraction the Behr Range for Soxhlet Extraction
Extraction The behr Range for Soxhlet Extraction The Soxhlet method is the standard extraction method. behr equipment for Soxhlet extraction meets the most varying requirements of daily work in a laboratory. practical stand for holding the condensers securely between extractions extractor sizes from 30 ml to 2000 ml compact systems with a locator for 1 sample multi-sample extractors with locators for 4 or 6 samples extractors with a specially developed siphon tube ensure a uniform extraction process at all sample positions the use of extractors with a stopcock does away with the need for any additional distillation after extraction Condensers with screwed- on tubing nipples Hydrolysis units (1, 4 or 6 sample places) for the acid digestion preceeding the extraction when determining the total fat content according to Weibull and Stoldt. Complete compact extraction systems Complete compact extraction systems with stand, heating module, mount, hoses and glass- ware (round-bottom reaction fl ask, extractor, Dimroth condenser for extraction). Infi nitely variable heating regulation. After the extraction cycle the compact systems drain the solvent through a drain cock at the extractor directly into the reservoir. KEX 100 F Compact systems for the Soxhlet extraction Model Article description 230 V~ Art.-No. 115 V~ Art.-No. KEX 30 Compact system for the 30-ml extraction 80 48 00520 80 48 005205 KEX 100 Compact system for the 100-ml extraction 80 48 00521 80 48 005215 KEX 250 Compact system for the 250-ml extraction 80 48 00540 80 48 005405 KEX 500 Compact system for the 500-ml extraction 80 48 00542 80 48 005425 18 Extraction Compact systems for the Soxhlet extraction Glass compositions Model Article description 230 V~ Art.-No. -

Evaluation of Extraction Methods for Recovery of Fatty Acids from Marine Products
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Universidade do Algarve Master thesis of EMQAL project Evaluation of Extraction Methods for Recovery of Fatty Acids from Marine Products Liping Xiao Supervisor: Svein Are Mjøs, Nofima Ingredients, Bjørn Grung, University of Bergen February 2010 Abstract The extraction efficiency of Soxhlet, acid hydrolysis and Bligh and Dyer were evaluated by using direct methylation on extracts and residues for calculating the mass balance of fatty acids for eight marine powders (fishmeals, krillmeals, cod filet, salmon filet and herring roe). The results show that Soxhlet gave lowest extracted fatty acid content, especially for the samples which contain a high amount of phospholipid. Acid hydrolysis and Bligh and Dyer extract gave comparable extracted fatty acid contents with direct methylation. The mass balance of fatty acids in extract and residue is close to 100% for the three extraction methods which indicate that fatty acid was not lost during the extraction procedures. The difference of extracted fatty acids is mainly due to the different extracting efficiency. The gravimetric lipid has limited correlation with total fatty acids, especially for Soxhlet. Analyses of the fatty acid profiles showed that the Soxhlet extracts were different from the others. Extracts from the acid hydrolysis and Bligh and Dyer methods had similar fatty acid profiles as the direct methylation method. The precision of fatty acid analysis by direct methylation method for marine powders were also validated. The coefficient of variation was 5.11% for solid samples and 1.21% for liquid sample. Key words: direct methylation, one-step methylation, fatty acids, Soxhlet, acid hydrolysis, Bligh and Dyer Table of Contents List of Abbreviations………………………………………………………….