Stagewise Processes Laboratory Handbook
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Solvent Extraction Instruments Brochure
ROT-X-TRACT SERİES Solvent Extraction Instruments PRODUCT BROCHURE ELEMENTEL ANALİTİK ve BİO TEKNOLOJİK SİSTEMLER | Folkart Towers - Adalet Mah. Manas Blv. No: 39/3408 Bayraklı/IZMIR, TURKEY, PHONE-FAX: +90 232 472 17 11 • [email protected] • www.elementel.com 2 Content www.elementel.com CONTENT ORGANOMATION SOLVENT EXTRACTORS OVERVIEW ..................................................................................... 3 ROT-X-TRACT-S SERIES ROTARY SOLID-LIQUID SOXHLET EXTRACTORS ........................................................ 5 ROT-X-TRACT-LC SERIES CORNING ACCELERATED ONE-STEP LIQUID-LIQUID EXTRACTORS ....................... 7 ROT-X-TRACT SOLVENT EXTRACTION INSTRUMENTS ACCESSORIES AND REPLACEMENT PARTS ............ 9 Organomation Solvent Extraction Instruments Overview 3 www.elementel.com Organomation Solvent Extractors Overview You are in the lab and need to run multiple extractions at once. First you set the Soxhlet extractors up on individual heating mantles or baths. Next, tubing is individually attached to each of the condensers. There are dozens of tubes to deal with, and multiple heating mantles to monitor. The problem with this arrangement is that in the end, you have spent more time assembling and monitoring your apparatuses than performing the actual chemistry. To conserve valuable bench space, all samples are arranged in a circle and the instrument rotates allowing each sample to be accessed from the front. Individual condensers are connected to the centrally located water manifold through quick disconnect fittings. -

Method 23 Determination of Polychlorinated Dibenzo-P-Dioxins
Method 23 8/3/2017 While we have taken steps to ensure the accuracy of this Internet version of the document, it is not the official version. To see a complete version including any recent edits, visit: https://www.ecfr.gov/cgi-bin/ECFR?page=browse and search under Title 40, Protection of Environment. METHOD 23—DETERMINATION OF POLYCHLORINATED DIBENZO-P-DIOXINS AND POLYCHLORINATED DIBENZOFURANS FROM STATIONARY SOURCES 1. Applicability and Principle 1.1 Applicability. This method is applicable to the determination of polychlorinated dibenzo-p- dioxins (PCDD's) and polychlorinated dibenzofurans (PCDF's) from stationary sources. 1.2 Principle. A sample is withdrawn from the gas stream isokinetically and collected in the sample probe, on a glass fiber filter, and on a packed column of adsorbent material. The sample cannot be separated into a particle vapor fraction. The PCDD's and PCDF's are extracted from the sample, separated by high resolution gas chromatography, and measured by high resolution mass spectrometry. 2. Apparatus 2.1 Sampling. A schematic of the sampling train used in this method is shown in Figure 23-1. Sealing greases may not be used in assembling the train. The train is identical to that described in section 2.1 of Method 5 of this appendix with the following additions: Method 23 8/3/2017 2.1.1 Nozzle. The nozzle shall be made of nickel, nickel-plated stainless steel, quartz, or borosilicate glass. 2.1.2 Sample Transfer Lines. The sample transfer lines, if needed, shall be heat traced, heavy 1 1 walled TFE ( ⁄2 in. -

Soxhlet Extraction of Solid Materials: an Outdated Technique with a Promising Innovative Future
Analytica Chimica Acta 369 (1998) 1±10 Soxhlet extraction of solid materials: an outdated technique with a promising innovative future M.D. Luque de Castro*, L.E. GarcõÂa-Ayuso Department of Analytical Chemistry, Faculty of Sciences, University of CoÂrdoba, E-14004 CoÂrdoba, Spain Received 8 December 1997; received in revised form 23 March 1998; accepted 30 March 1998 Abstract An overview of the evolution of Soxhlet extraction of solid materials and its comparison with the performance of other conventional and new extraction techniques is presented. First, a discussion on both conventional Soxhlet as compared with other conventional extraction techniques and some minor improvements of the former for speci®c applications is done. Secondly, a critical comparison of conventional Soxhlet with the new extraction techniques such as supercritical ¯uid extraction, microwave-assisted processes and microwave-assisted solvent extraction shows the reasons why major, recent improvements of this technique (namely Soxtec1 System HT, Soxwave-100 and focused microwave-assisted Soxhlet extraction) have been proposed, aimed at overcoming most of the shortcomings of conventional Soxhlet and converting it into an updated tool for leaching which competes advantageously with the most recent alternatives in the extraction ®eld. # 1998 Elsevier Science B.V. All rights reserved. Keywords: Updated Soxhlet; Review 1. Introduction physicochemical terminology, as leaching or lixivia- tion, is one of the oldest ways of solid sample pre- Sample pretreatment is often one of the most time treatment. Among the techniques used for consuming steps of the analytical process, particularly implementation of this step, Soxhlet has been the when solid samples are involved. The search for leaching technique mostly used for a long time. -

Alembic Pot Still
ALEMBIC POT STILL INSTRUCTION MANUAL CAN BE USED WITH THE GRAINFATHER OR T500 BOILER SAFETY Warning: This system produces a highly flammable liquid. PRECAUTION: • Always use the Alembic Pot Still System in a room with adequate ventilation. • Never leave the Alembic Pot Still system unattended when operating. • Keep the Alembic Pot Still system away from all sources of ignition, including smoking, sparks, heat, and open flames. • Ensure all other equipment near to the Alembic Pot Still system or the alcohol is earthed. • A fire extinguishing media suitable for alcohol should be kept nearby. This can be water fog, fine water spray, foam, dry powder, carbon dioxide, sand or dolomite. • Do not boil dry. In the event the still is boiled dry, reset the cutout button under the base of the still. In the very unlikely event this cutout fails, a fusible link gives an added protection. IN CASE OF SPILLAGE: • Shut off all possible sources of ignition. • Clean up spills immediately using cloth, paper towels or other absorbent materials such as soil, sand or other inert material. • Collect, seal and dispose accordingly • Mop area with excess water. CONTENTS Important points before getting started ............................................................................... 3 Preparing the Alembic Pot Still ................................................................................................. 5 Distilling a Whiskey, Rum or Brandy .......................................................................................7 Distilling neutral -

Minimum Reflux in Liquid–Liquid Extraction
17th European Symposium on Computer Aided Process Engineering – ESCAPE17 V. Plesu and P.S. Agachi (Editors) © 2007 Elsevier B.V. All rights reserved. 1 Minimum Reflux in Liquid–Liquid Extraction Santanu Bandyopadhyaya and Calin-Cristian Cormosb aEnergy Systems Engineering, Indian Institute of Technology, Bombay, Powai, Mumbai 400 076, India, E-mail: [email protected] bDepartment of Chemical Engineering, Faculty of Chemistry and Chemical Engineering, University "Babeş-Bolyai", Arany Janos 11, Cluj-Napoca 400028, Romania, E-mail: [email protected] Abstract In a simple countercurrent arrangement of different stages of liquid–liquid extraction operation, the richest extract leaving the operation is in equilibrium with the feed. However, by using reflux it is possible to enrich the extract further. A simple counter current liquid–liquid extraction operation with reflux is analogous in its essentials to distillation. In this paper, the method based on Invariant Rectifying-Stripping (IRS) curves, originally proposed to calculate minimum reflux and minimum energy requirement in distillation, has been extended to liquid–liquid extraction. The equivalent IRS curves for a ternary liquid–liquid extraction predicts the feed location in the counter current process. It also predicts the minimum reflux requirement for a given separation and minimum amount of solvent required. Keywords: liquid extraction, IRS curves, feed location, minimum solvent. 1. Introduction Liquid–liquid extraction is a separation process that takes advantage of the distribution of a substance between two insoluble liquids phases [1]. Feed to be separated is mixed with extracting solvent to produce a solvent-rich phase, called extract, and a solvent-lean phase, called raffinate. -

Reflux Condensation in Narrow Rectangular Channels with Perforated Fins Nadia Souidi, André Bontemps
Reflux condensation in narrow rectangular channels with perforated fins Nadia Souidi, André Bontemps To cite this version: Nadia Souidi, André Bontemps. Reflux condensation in narrow rectangular channels with perforated fins. Applied Thermal Engineering, Elsevier, 2003, 23, pp.871-891. 10.1016/S1359-4311(03)00021-8. hal-00184135 HAL Id: hal-00184135 https://hal.archives-ouvertes.fr/hal-00184135 Submitted on 19 Feb 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Reflux condensation in narrow rectangular channels with perforated fins N. Souidi a, A. Bontemps b,* a GRETh-CEA Grenoble, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France b LEGI-GRETh, Universitee Joseph Fourier, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France Reflux condensation is an industrial process that aims to reduce the content of the less volatile com- ponent or to eliminate the non-condensable phase of a vapour mixture, by the means of separation. Separation consists in condensing the less volatile phase and to recover the condensate while simulta- neously, the non-condensable species are recuperated at the top of the system. Compact plate-fin heat exchangers can be used in gas separation processes. -

Hyoscyami Semen (Unprocessed)(Unprocessed)
HyoscyamiHyoscyami Semen (unprocessed)(unprocessed) B CC 0.50.5 mm mm AA 11 mm FigureFigure 1 1 A A photographphotograph of Hyoscyami Semen (unprocessed)(unprocessed) A.A. Hyoscyami Hyoscyami SemenSemen (unprocessed)(unprocessed) B. Magnified image of seeds B. Magnified image of seeds C. Magnified image of longitudinal section of seeds C. Magnified image of longitudinal section of seeds 175 175 Hyoscyami Semen (unprocessed) Hyoscyami Semen (unprocessed) 1. NAMES Powder Exotesta cells scattered or in groups, yellow or greyish-yellowish brown, polygonal or elongated Official Name: Hyoscyami Semen (unprocessed) polygonal in surface view, 75-311 µm long, 25-123 µm in diameter, anticlinal walls thickened, Chinese Name: undulant, with distinct striations, containing yellowish-brown contents; bright yellowish-white under the polarized microscope. Endosperm cells polygonal or subrounded in surface view, cell Chinese Phonetic Name: Tianxianzi (Sheng) walls slightly thickened, containing aleurone grains and oil droplets. Cotyledon cells colourless, subpolygonal, with thin walls, containing oil droplets (Fig. 3). 2. SOURCE Hyoscyami Semen (unprocessed) is the unprocessed dried ripe seed of Hyoscyamus niger L. (Solanaceae). The fruit is collected in summer and autumn when the pericarp turns yellow, exposed to the sun, afterwards the seeds tapped out, pericarp and stalk removed, then dried under the sun to obtain Hyoscyami Semen (unprocessed). 3. DESCRIPTION Flattened-subreniform to flattened-ovoid, about 1 mm in diameter. Externally brownish-yellow or greyish-yellow, with fine and dense reticulate striations, and a pitted hilum located at the slightly acute end. Cut surface greyish-white, oily, containing endosperm, embryo curved. Odour slight (Fig. 1). 4. IDENTIFICATION 4.1 Microscopic Identification (Appendix III) Transverse section Exotesta cells with irregularly undulant protuberances, apex of the protuberances tapering or blunt, cell walls with transparent striations. -

Building a Home Distillation Apparatus
BUILDING A HOME DISTILLATION APPARATUS A Step by Step Guide Building a Home Distillation Apparatus i BUILDING A HOME DISTILLATION APPARATUS Foreword The pages that follow contain a step-by-step guide to building a relatively sophisticated distillation apparatus from commonly available materials, using simple tools, and at a cost of under $100 USD. The information contained on this site is directed at anyone who may want to know more about the subject: students, hobbyists, tinkers, pure water enthusiasts, survivors, the curious, and perhaps even amateur wine and beer makers. Designing and building this apparatus is the only subject of this manual. You will find that it confines itself solely to those areas. It does not enter into the domains of fermentation, recipes for making mash, beer, wine or any other spirits. These areas are covered in detail in other readily available books and numerous web sites. The site contains two separate design plans for the stills. And while both can be used for a number of distillation tasks, it should be recognized that their designs have been optimized for the task of separating ethyl alcohol from a water-based mixture. Having said that, remember that the real purpose of this site is to educate and inform those of you who are interested in this subject. It is not to be construed in any fashion as an encouragement to break the law. If you believe the law is incorrect, please take the time to contact your representatives in government, cast your vote at the polls, write newsletters to the media, and in general, try to make the changes in a legal and democratic manner. -

Seed Oil Using Soxhlet Method
S.H. Mohd-SetaparMalaysian et al. Journal / Malaysian of Fundamental Journal of andFundamental Applied Sciences and Applied Vol.10, Sciences No.1 (2014)Vol.10, 1-6 No.1 (2014) 1-6 Extraction of rubber (Hevea brasiliensis) seed oil using soxhlet method S.H. Mohd-Setapar*, Lee Nian-Yian and N.S. Mohd-Sharif Centre of Lipid Engineering & Applied Research (CLEAR), Dept. of Chemical Eng., Fac. of Chemical Eng., Universiti Teknologi Malaysia 81310 Skudai, Johor, Malaysia. *Corresponding Author: [email protected] (S.H. Mohd-Setapar) Article history : ABSTRACT Received 4 January 2013 Revised 24 June 2013 Soxhlet extraction which is also known as solvent extraction refers to the preferential dissolution of oil by Accepted 1 July 2013 contacting oilseeds with a liquid solvent. This is the most efficient method to recover oil from oilseeds, Available online 1 August 2013 thus solvent extraction using hexane has been commercialized as a standard practice in today’s industry. In this study, soxhlet extraction had been used to extract the rubber seed oil which contains high GRAPHICAL ABSTRACT percentage of alpha-linolenic acid. In addition, the different solvents will be used for the extraction of rubber seed oil such as petroleum ether, n-hexane, ethanol and water to study the best solvent to extract the rubber seed oil so the maximum oil yield can be obtained. On the other hands, the natural resource, rubber belongs to the family of Euphorbiaceae, the genus is Hevea while the species of rubber is brasiliensis. Rubber (Hevea brasiliensis) seeds are abundant and wasted because they had not been used in any industry or applications in daily life. -

Batch Distillation of Spirits: Experimental Study and Simulation
Research article Received: 5 April 2018 Revised: 11 January 2019 Accepted: 15 January 2019 Published online in Wiley Online Library (wileyonlinelibrary.com) DOI 10.1002/jib.560 Batch distillation of spirits: experimental study and simulation of the behaviour of volatile aroma compounds Adrien Douady,1 Cristian Puentes,1 Pierre Awad1,2 and Martine Esteban-Decloux1* This paper focuses on the behaviour of volatile compounds during batch distillation of wine or low wine, in traditional Charentais copper stills, heated with a direct open flame at laboratory (600 L) and industrial (2500 L) scale. Sixty-nine volatile compounds plus ethanol were analysed during the low wine distillation in the 600 L alembic still. Forty-four were quantified and classified according to their concentration profile in the distillate over time and compared with previous studies. Based on the online re- cording of volume flow, density and temperature of the distillate with a Coriolis flowmeter, distillation was simulated with ProSim® BatchColumn software. Twenty-six volatile compounds were taken into account, using the coefficients of the ‘Non- Random Two Liquids’ model. The concentration profiles of 18 compounds were accurately represented, with slight differences in the maximum concentration for seven species together with a single compound that was poorly represented. The distribution of the volatile compounds in the four distillate fractions (heads, heart, seconds and tails) was well estimated by simulation. Fi- nally, data from wine and low wine distillations in the large-scale alembic still (2500 L) were correctly simulated, suggesting that it was possible to adjust the simulation parameters with the Coriolis flowmeter recording and represent the concentration pro- files of most of the quantifiable volatile compounds. -

Soxhlet Extraction Classic Fat Determination with Soxhlet Extraction After Hydrolysis
Soxhlet extraction Classic fat determination with Soxhlet extraction after hydrolysis Hydrolysis principle This acid digestion process dissolves both "free fats" as well as "bound fats" from the overall fat content. The fat is frequently naturally enclosed in the cell matrix of the food or fodder or chemically bound. In these cases, a hydrolysis step before extraction completely releases the fat. Hydrolysis-digestion units p. 18 The user filters the hydrolysate of the separated sample by using a glass sample tube filled with sand or Celite. The user then rinses the fatty filter residue with water in order to remove the acid. Filter equipment for hydrolysis p. 18 After drying, the filter residue is finally extracted. Soxhlet extraction systems p. 9 8 Soxhlet Complete single extraction units The standard extraction method is the Soxhlet method. behr apparatus for Soxhlet extractions fulfil all the various requirements in everyday laboratory practice. n Practical brackets for condensers and intermediate extraction pieces for safe storage between extractions n Extractor sizes from 30 ml to 1,000 ml n Compact apparatus with one sample position n Series extraction devices with 4, 6 or 8 sample positions n Extractors with specially developed siphon tubes (make: "Bröckerhoff") guarantee consistent extraction cycles across all sample positions n Extractors with taps remove the need for additional distillation after the extraction n Condensers with threaded fittings The behr hydrolysis units (4 or 6 sample positions) also enable acid digestion prior to extraction (determination of the total fat content according to Weibull and Stoldt). Complete single extraction units Complete single extraction units with base frame, heating device, bracket, tubes and glass apparatus (reaction flask, extractor, Dimroth condenser for extraction). -
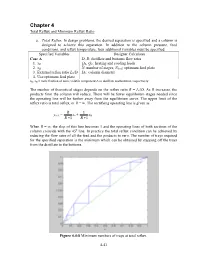
Minimum Reflux Ratio
Chapter 4 Total Reflux and Minimum Reflux Ratio a. Total Reflux . In design problems, the desired separation is specified and a column is designed to achieve this separation. In addition to the column pressure, feed conditions, and reflux temperature, four additional variables must be specified. Specified Variables Designer Calculates Case A D, B: distillate and bottoms flow rates 1. xD QR, QC: heating and cooling loads 2. xB N: number of stages, Nfeed : optimum feed plate 3. External reflux ratio L0/D DC: column diameter 4. Use optimum feed plate xD, xB = mole fraction of more volatile component A in distillate and bottoms, respectively The number of theoretical stages depends on the reflux ratio R = L0/D. As R increases, the products from the column will reduce. There will be fewer equilibrium stages needed since the operating line will be further away from the equilibrium curve. The upper limit of the reflux ratio is total reflux, or R = ∞. The rectifying operating line is given as R 1 yn+1 = xn + xD R +1 R +1 When R = ∞, the slop of this line becomes 1 and the operating lines of both sections of the column coincide with the 45 o line. In practice the total reflux condition can be achieved by reducing the flow rates of all the feed and the products to zero. The number of trays required for the specified separation is the minimum which can be obtained by stepping off the trays from the distillate to the bottoms. Figure 4.4-8 Minimum numbers of trays at total reflux.