Simulating Plasmon Enhancement of Optical Properties in Hybrid Metal-Organic Nanoparticles
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Novel Magnetic Materials Based on Macrocyclic Ligands: Towards High Relaxivity Contrast Agents and Mononuclear Single-Molecule Magnets
Novel Magnetic Materials Based on Macrocyclic Ligands: Towards High Relaxivity Contrast Agents and Mononuclear Single-Molecule Magnets Emma Stares A thesis submitted to the Department of Chemistry in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Supervised by Professor Melanie Pilkington Brock University St. Catharines Ontario, Canada August 2015 © Emma Stares, 2015 Abstract The preparation and characterization of coordination complexes of Schiff-base and crown ether macrocycles is presented, for application as contrast agents for magnetic resonance imaging, Project 1; and single-molecule magnets (SMMs), Projects 2 and 3. II III In Project 1, a family of eight Mn and Gd complexes of N3X2 (X = NH, O) and N3O3 Schiff-base macrocycles were synthesized, characterized, and evaluated as potential contrast agents for MRI. In vitro and in vivo (rodent) studies indicate that the studied complexes display efficient contrast behaviour, negligible toxicity, and rapid excretion. III In Project 2, Dy complexes of Schiff-base macrocycles were prepared with a view to developing a new family of mononuclear Ln-SMMs with pseudo-D5h geometries. Each complex displayed slow relaxation of magnetization, with magnetically-derived energy barriers in the range Ueff = 4 – 24 K. In Project 3, coordination complexes of selected later lanthanides with various crown ether ligands were synthesized. Two families of complexes were structurally and magnetically analyzed: ‘axial’ or sandwich-type complexes based on 12-crown-4 and 15- crown-5; and ‘equatorial’ complexes based on 18-crown-6. Magnetic data are supported by ab initio calculations and luminescence measurements. Significantly, the first mononuclear Ln-SMM prepared from a crown ether ligand is described. -

Chloranilate Frameworks Katherine Bondaruk and Carol Hua* School of Chemistry, the University of Melbourne, Parkville, Victoria, 3010, Australia
Electronic Supplementary Information for: The Effect of Counterions on the Formation and Structures of Ce(III) and Er(III) Chloranilate Frameworks Katherine Bondaruk and Carol Hua* School of Chemistry, The University of Melbourne, Parkville, Victoria, 3010, Australia. *E-mail: [email protected] Table of Contents Table S1. Crystallographic parameters for the 2D (6,3) honeycomb frameworks obtained in this study..S2 Table S2. Crystallographic parameters for the 3D diamond frameworks obtained in this study ...............S3 + Table S3. Crystallographic parameters for frameworks containing PPh3Me obtained in this study.........S4 Table S4. Analysis of the possible coordination geometries using the SHAPE program for the 9- coordinate Ce(III) containing structures, compounds 1, 3 and 5.................................................................S5 Table S5. Analysis of the possible coordination geometries using the SHAPE program for the 8- coordinate Er(III) containing structures, compounds 2, 4 and 7. Where more than one unique Er(III) exists, the analysis is shown for each of the Er(III) centres. .......................................................................S6 Table S6. Analysis of the possible coordination geometries using the SHAPE program for the 9- coordinate Ce(III) centres in compound 6...................................................................................................S7 1-2 Figure S1. The (6,3) [Ce2(can)3(H2O)6] network in [Ce2(can)3(H2O)6]·12H2O ......................................S8 Figure S2. The 3D anionic framework, (DPMP)[Er(can)2] (4) with dia topology. ...................................S8 + + Figure S3. The calculated and experimental powder XRD patterns of 1 with Bu4N and Me4N cations. S9 + + Figure S4. The calculated and experimental powder XRD patterns of 2 with Bu4N and Me4N cations S9 Figure S5. The experimental and calculated powder XRD patterns of 3 .................................................S10 Figure S6. -

Volume 75 (2019)
Acta Cryst. (2019). B75, doi:10.1107/S2052520619010047 Supporting information Volume 75 (2019) Supporting information for article: Lanthanide coordination polymers based on designed bifunctional 2-(2,2′:6′,2″-terpyridin-4′-yl)benzenesulfonate ligand: syntheses, structural diversity and highly tunable emission Yi-Chen Hu, Chao Bai, Huai-Ming Hu, Chuan-Ti Li, Tian-Hua Zhang and Weisheng Liu Acta Cryst. (2019). B75, doi:10.1107/S2052520619010047 Supporting information, sup-1 Table S1 Continuous Shape Measures (CShMs) of the coordination geometry for Eu3+ ions in 1- Eu. Label Symmetry Shape 1-Eu EP-9 D9h Enneagon 33.439 OPY-9 C8v Octagonal pyramid 22.561 HBPY-9 D7h Heptagonal bipyramid 15.666 JTC-9 C3v Johnson triangular cupola J3 15.263 JCCU-9 C4v Capped cube J8 10.053 CCU-9 C4v Spherical-relaxed capped cube 9.010 JCSAPR-9 C4v Capped square antiprism J10 2.787 CSAPR-9 C4v Spherical capped square antiprism 1.930 JTCTPR-9 D3h Tricapped trigonal prism J51 3.621 TCTPR-9 D3h Spherical tricapped trigonal prism 2.612 JTDIC-9 C3v Tridiminished icosahedron J63 12.541 HH-9 C2v Hula-hoop 9.076 MFF-9 Cs Muffin 1.659 Acta Cryst. (2019). B75, doi:10.1107/S2052520619010047 Supporting information, sup-2 Table S2 Continuous Shape Measures (CShMs) of the coordination geometry for Ln3+ ions in 2- Er, 4-Tb, and 6-Eu. Label Symmetry Shape 2-Er 4-Tb 6-Eu Er1 Er2 OP-8 D8h Octagon 31.606 31.785 32.793 31.386 HPY-8 C7v Heptagonal pyramid 23.708 24.442 23.407 23.932 HBPY-8 D6h Hexagonal bipyramid 17.013 13.083 12.757 15.881 CU-8 Oh Cube 11.278 11.664 8.749 11.848 -

Supporting Information
Supporting Information Exploring the Slow Magnetic Relaxation of a Family of Photoluminescent 3D Lanthanide-Organic Frameworks Based on Dicarboxylate Ligands Itziar Oyarzabal, Sara Rojas, Ana D. Parejo, Alfonso Salinas-Castillo, José Ángel García, José M. Seco, Javier Cepeda and Antonio Rodríguez-Diéguez Index: 1. Powder X-ray Diffraction 2. Additional structural data 3. Interpretation of void content from SQUEEZE analysis 4. Continuous Shape Measures Calculations 5. Magnetic Properties 6. Luminescence Properties 1 S1. Powder X-ray Diffraction Table S1. Data of pattern-matching refinement of compound 2. Space group P-1 a (Å) 10.87(2) b(Å) 11.11(2) c(Å) 13.26(3) α (°) 105.28(2) β (°) 94.22(3) γ (°) 93.65(2) V/ Å3 1534(3) Figure S1. Pattern-matching analysis and crystalline parameters of the polycrystalline sample of compound 2. Table S2. Data of pattern-matching refinement of compound 3. Space group P-1 a (Å) 10.85(2) b(Å) 11.04(4) c(Å) 13.21(2) α (°) 105.01(2) β (°) 94.90(4) γ (°) 93.90(4) V/ Å3 1516(5) Figure S2. Pattern-matching analysis and crystalline parameters of the polycrystalline sample of compound 3. 2 Table S3. Data of pattern-matching refinement of compound 4. Space group P-1 a (Å) 10.86(2) b(Å) 11.03(4) c(Å) 13.20(2) α (°) 105.20(2) β (°) 94.38(4) γ (°) 93.75(4) V/ Å3 1515(5) Figure S3. Pattern-matching analysis and crystalline parameters of the polycrystalline sample of compound 4. 3 S2. -

Metallaheteroboranes Containing Group 16 Elements: an Experimental and Theoretical Study
Journal of Organometallic Chemistry 883 (2019) 71e77 Contents lists available at ScienceDirect Journal of Organometallic Chemistry journal homepage: www.elsevier.com/locate/jorganchem Metallaheteroboranes containing group 16 elements: An experimental and theoretical study * Moulika Bhattacharyya, Rini Prakash, R. Jagan, Sundargopal Ghosh Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India article info abstract Article history: Thermolysis of diphenyl dichalcogenides, Ph2E2 (E ¼ S, Se and Te) with an insitu generated intermediate, Received 19 November 2018 5 obtained from the reaction of [Cp*WCl4] (Cp* ¼ h -C5Me5) with [LiBH4$THF], yielded new tung- Received in revised form staheteroboranes, [(Cp*W)2(m-EPh)(m3-E)(m-H)(B3H2EPh)], 1 and 2 (1:E¼ S; 2:E¼ Se), and [(Cp*W)2(m- 9 January 2019 TePh)B H (m-H) ], 4. Formation of compounds 1 and 2 substantiate the evidence of [(Cp*W) B H ], Accepted 11 January 2019 5 5 3 2 4 10 which supports our previous studies. Compound 4 has a capped octahedral geometry that is unique due Available online 17 January 2019 to the presence of three W-H-B bridging hydrogens in a closo cluster. Alternatively, the shape of cluster 4 can be defined as oblatoarachno that can be derived from a heptagonal bipyramid. All the compounds Keywords: 1 11 1 13 1 Oblatoarachno have been characterized by mass spectrometry, H, B{ H}, IR and C{ H} NMR spectroscopy in solution Metallaheteroboranes and the structural architectures of 1 and 4 were unequivocally established by X-ray crystallographic Tungsten analysis. The density functional theory calculations also yielded geometries in close agreement with the Diphenyl dichalcogenide observed structures. -

Biomimetics Scrivener Publishing 100 Cummings Center, Suite 541J Beverly, MA 01915-6106
Biomimetics Scrivener Publishing 100 Cummings Center, Suite 541J Beverly, MA 01915-6106 Biomedical Science, Engineering, and Technology The book series seeks to compile all the aspects of biomedi- cal science, engineering and technology from fundamental principles to current advances in translational medicine. It covers a wide range of the most important topics including, but not limited to, biomedical materials, biodevices and biosys- tems, bioengineering, micro and nanotechnology, biotechnology, biomolecules, bioimaging, cell technology, stem cell engineering and biology, gene therapy, drug delivery, tissue engineering and regeneration, and clinical medicine. Series Editor: Murugan Ramalingam, Centre for Stem Cell Research Christian Medical College Bagayam Campus Vellore-632002, Tamilnadu, India E-mail: [email protected] Publishers at Scrivener Martin Scrivener ([email protected]) Phillip Carmical ([email protected]) Biomimetics Advancing Nanobiomaterials and Tissue Engineering Edited by Murugan Ramalingam, Xiumei Wang, Guoping Chen, Peter Ma, and Fu-Zhai Cui Copyright © 2013 by Scrivener Publishing LLC. All rights reserved. Co-published by John Wiley & Sons, Inc. Hoboken, New Jersey, and Scrivener Publishing LLC, Salem, Massachusetts. Published simultaneously in Canada. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning, or other - wise, except as permitted under Section 107 or 108 of the 1976 United States Copyright Act, without either the prior written permission of the Publisher, or authorization through payment of the appropriate per-copy fee to the Copyright Clearance Center, Inc., 222 Rosewood Drive, Danvers, MA 01923, (978) 750-8400, fax (978) 750-4470, or on the web at www.copyright.com. -

ACS Style Guide
➤ ➤ ➤ ➤ ➤ The ACS Style Guide ➤ ➤ ➤ ➤ ➤ THIRD EDITION The ACS Style Guide Effective Communication of Scientific Information Anne M. Coghill Lorrin R. Garson Editors AMERICAN CHEMICAL SOCIETY Washington, DC OXFORD UNIVERSITY PRESS New York Oxford 2006 Oxford University Press Oxford New York Athens Auckland Bangkok Bogotá Buenos Aires Calcutta Cape Town Chennai Dar es Salaam Delhi Florence Hong Kong Istanbul Karachi Kuala Lumpur Madrid Melbourne Mexico City Mumbai Nairobi Paris São Paulo Singapore Taipei Tokyo Toronto Warsaw and associated companies in Berlin Idaban Copyright © 2006 by the American Chemical Society, Washington, DC Developed and distributed in partnership by the American Chemical Society and Oxford University Press Published by Oxford University Press, Inc. 198 Madison Avenue, New York, NY 10016 Oxford is a registered trademark of Oxford University Press All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, or otherwise, without the prior permission of the American Chemical Society. Library of Congress Cataloging-in-Publication Data The ACS style guide : effective communication of scientific information.—3rd ed. / Anne M. Coghill [and] Lorrin R. Garson, editors. p. cm. Includes bibliographical references and index. ISBN-13: 978-0-8412-3999-9 (cloth : alk. paper) 1. Chemical literature—Authorship—Handbooks, manuals, etc. 2. Scientific literature— Authorship—Handbooks, manuals, etc. 3. English language—Style—Handbooks, manuals, etc. 4. Authorship—Style manuals. I. Coghill, Anne M. II. Garson, Lorrin R. III. American Chemical Society QD8.5.A25 2006 808'.06654—dc22 2006040668 1 3 5 7 9 8 6 4 2 Printed in the United States of America on acid-free paper ➤ ➤ ➤ ➤ ➤ Contents Foreword. -

Synthesis and Optical Properties of Gold Nanodecahedra with Size Control**
COMMUNICATIONS DOI: 10.1002/adma.200600475 Synthesis and Optical Properties of Gold Nanodecahedra with Size Control** By Ana Sánchez-Iglesias, Isabel Pastoriza-Santos, Jorge Pérez-Juste, Benito Rodríguez-González, F. Javier García de Abajo, and Luis M. Liz-Marzán* The synthesis of nanoparticles with a tight control on their this reason they do not qualify as Platonic nanocrystals, but size and shape is a requirement for the achievement of many can rather be classified as Johnson structures. These decahe- nanotechnology goals, since these are the main parameters dral particles have a lower symmetry (D5h) than Platonic sol- determining the material properties at the nanoscale. Among ids, but still display a striking beauty and attractive optical the many different materials currently under investigation, properties that are strongly influenced by particle size. Al- semiconductor and metal nanoparticles are extremely attrac- though the geometry (and thus the aspect ratio) is precisely tive because of the possibility of controlling their electronic the same for particles with different sizes, clear changes in the and optical properties through tailoring size and shape during optical responses have been observed and modeled through a synthesis. For instance, the presence of sharp edges or tips has boundary element method (BEM) for bicones. been shown to increase electric-field enhancement,[1] which is The synthesis is based on our previous work where N,N-di- important for applications involving metal nanoparticles as methylformamide (DMF) was used as both solvent and reduc- sensors. Additionally, nanoparticle morphology will ultimately ing agent[16] to generate silver nanoparticles of various shapes, determine the way in which nanoparticles can be assembled. -
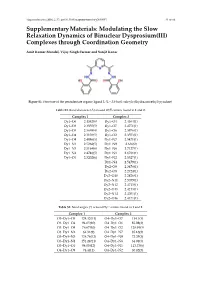
Supplementary Materials: Modulating the Slow Relaxation Dynamics of Binuclear Dysprosium(III) Complexes Through Coordination Geometry
Magnetochemistry 2016, 2, 35; doi:10.3390/magnetochemistry2030035 S1 of S9 Supplementary Materials: Modulating the Slow Relaxation Dynamics of Binuclear Dysprosium(III) Complexes through Coordination Geometry Amit Kumar Mondal, Vijay Singh Parmar and Sanjit Konar Figure S1. Structure of the pentadentate organic ligand L (L = 2,6-bis(1-salicyloylhydrazonoethyl) pyridine). Table S1. Bond distances (Å) around DyIII centers found in 1 and 2. Complex 1 Complex 2 Dy1–O6 2.4382(6) Dy1–O4 2.4364(1) Dy1–O3 2.3855(5) Dy1–O7 2.4771(1) Dy1–O5 2.5639(4) Dy1–O6 2.3976(1) Dy1–O4 2.3103(5) Dy1–O2 2.3553(1) Dy1–O9 2.4086(5) Dy1–N7 2.5471(1) Dy1–N1 2.5268(5) Dy1–N9 2.636(2) Dy1–N3 2.5164(6) Dy1–N6 2.7127(1) Dy1–N4 2.4746(5) Dy1–N1 2.6781(1) Dy1–O1 2.3202(6) Dy1–N2 2.5527(1) Dy1–N4 2.5479(1) Dy2–O9 2.3478(1) Dy2–O8 2.2724(1) Dy2–O10 2.2826(1) Dy2–N11 2.5059(1) Dy2–N12 2.4713(1) Dy2–O13 2.4213(1) Dy2–N14 2.4701(1) Dy2–O16 2.4171(1) Table S2. Bond angles (°) around DyIII centers found in 1 and 2. Complex 1 Complex 2 O3–Dy1–O5 124.32(11) O4–Dy1–O7 114.1(3) O3–Dy1–O4 94.65(10) O4–Dy1–O6 85.08(3) O3–Dy1–O9 76.67(10) O4–Dy1–O2 128.91(3) O3–Dy1–N1 64.31(9) O4–Dy1–N7 85.16(3) O3–Dy1–N3 124.76(12) O4–Dy1–N9 72.29(3) O3–Dy1–N4 151.24(11) O4–Dy1–N6 64.04(3) O3–Dy1–O1 94.05(12) O4–Dy1–N1 113.73(6) O4–Dy1–O9 74.4(11) O4–Dy1–N2 60.82(3) Magnetochemistry 2016, 2, 35; doi:10.3390/magnetochemistry2030035 S2 of S9 Table S2. -

Asymmetric Dinuclear Lanthanide(III) Complexes from the Use Of
Supplementary Materials Asymmetric Dinuclear Lanthanide(III) Complexes from the Use of a Ligand Derived from 2- Acetylpyridine and Picolinoylhydrazide: Synthetic, Structural and Magnetic Studies† Diamantoula Maniaki 1, Panagiota S. Perlepe 2,3, Evangelos Pilichos 1, Sotirios Christodoulou 4, Mathieu Rouzières 2, Pierre Dechambenoit 2,*, Rodolphe Clérac 2,* and Spyros P. Perlepes 1,5,* 1 Department of Chemistry, University of Patras, 265 04 Patras, Greece; [email protected] (D.M.); [email protected] (E.P.) 2 Univ. Bordeaux, CNRS, Centre de Recherche Paul Pascal, UMR 5031, 336 00, Pessac, France; [email protected] (P.S.P.); [email protected] (M.R.) 3 Univ. Bordeaux, CNRS, ICMCB, UMR 5026, 336 00, Pessac, France 4 ICFO-Institut de Ciencies Fotoniques, The Barcelona Institute of Nanoscience and Nanotechnology, Castelldefels, 088 60 Barcelona, Spain; [email protected] (S.C.) 5 Foundation for Research and Technology-Hellas (FORTH), Institute of Chemical Engineering Sciences (ICE-HT), Platani, P.O. Box 1414, 265 04 Patras, Greece * Correspondence: [email protected] (P.D.); [email protected] (R.C.); [email protected] (S.P.P.); Tel: +33-556845671 (P.D.); +33-556845650 (R.C.); +30-2610-996730 (S.P.P.) † This article is dedicated to the memory of Professor Kyriakos Riganakos, an excellent academician, a great food chemistry scientist and a precious friend. Received: 25 May 2020; Accepted: 5 July 2020; Published: date Molecules 2020, 25, 3153; doi:10.3390/molecules25143153 www.mdpi.com/journal/molecules Molecules 2020, 25, 3153 2 of 10 Figure S1. Crystal structure of [Gd2(NO3)4(L)2(H2O)] as found in 1∙2MeOH∙2H2O at 120 K. -
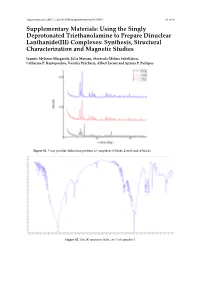
Complexes: Synthesis, Structural Characterization and Magnetic Studies
Magnetochemistry 2017, 3, doi:10.3390/magnetochemistry3010005 S1 of S6 Supplementary Materials: Using the Singly Deprotonated Triethanolamine to Prepare Dinuclear Lanthanide(III) Complexes: Synthesis, Structural Characterization and Magnetic Studies Ioannis Mylonas-Margaritis, Julia Mayans, Stavroula-Melina Sakellakou, Catherine P. Raptopoulou, Vassilis Psycharis, Albert Escuer and Spyros P. Perlepes Figure S1. X-ray powder diffraction patterns of complexes 3 (blue), 2 (red) and 4 (black). Figure S2. The IR spectrum (KBr, cm−1) of complex 3. Magnetochemistry 2017, 3, 5 S2 of S6 Figure S3. The IR spectrum (liquid between CsI disks, cm−1) of the free teaH3 ligand. Figure S4. Partially labeled plot of the molecule [Pr2(NO3)4(teaH2)2] that is present in the structure of 1∙2MeOH. Symmetry operation used to generate equivalent atoms: (΄) –x + 1, −y, −z + 1. Only the H atoms at O2 and O3 (and their centrosymmetric equivalents) are shown. Magnetochemistry 2017, 3, 5 S3 of S6 Figure S5. Partially labeled plot of the molecule [Gd2(NO3)4(teaH2)2] that is present in the structure of 2∙2MeOH. Symmetry operation used to generate equivalent atoms: (΄) –x + 1, −y, −z + 1. Only the H atoms at O2 and O3 (and their centrosymmetric equivalents) are shown. Figure S6. The spherical capped square antiprismatic coordination geometry of Dy1 in the structure of 4∙2MeOH. The plotted polyhedron is the ideal, best-fit polyhedron using the program SHAPE. Primed and unprimed atoms are related by the symmetry operation (΄) –x + 1, −y, −z + 1. Magnetochemistry 2017, 3, 5 S4 of S6 GdIII Figure S7. The Johnson tricapped trigonal prismatic coordination geometry of Gd1 in the structure of 2∙2MeOH. -

Abstract Shape Synthesis from Linear Combinations of Clelia Curves
The 8th ACM/EG Expressive Symposium EXPRESSIVE 2019 C. Kaplan, A. Forbes, and S. DiVerdi (Editors) Abstract Shape Synthesis From Linear Combinations of Clelia Curves L. Putnam, S. Todd and W. Latham Computing, Goldsmiths, University of London, United Kingdom Abstract This article outlines several families of shapes that can be produced from a linear combination of Clelia curves. We present parameters required to generate a single curve that traces out a large variety of shapes with controllable axial symmetries. Several families of shapes emerge from the equation that provide a productive means by which to explore the parameter space. The mathematics involves only arithmetic and trigonometry making it accessible to those with only the most basic mathematical background. We outline formulas for producing basic shapes, such as cones, cylinders, and tori, as well as more complex fami- lies of shapes having non-trivial symmetries. This work is of interest to computational artists and designers as the curves can be constrained to exhibit specific types of shape motifs while still permitting a liberal amount of room for exploring variations on those shapes. CCS Concepts • Computing methodologies ! Parametric curve and surface models; • Applied computing ! Media arts; 1. Introduction curves [Cha15, Gai05] edge closer to balancing constraint and freedom. There is a large repertoire of mathematical systems and algorithms capable of generating rich and complex non-figurative graphics We draw inspiration from specific artistic works such as Ernst [Whi80,