Bet1 CRISPR Activation Plasmid (M): Sc-419320-ACT
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Comparative Analysis of Human Chromosome 7Q21 and Mouse
Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Comparative analysis of human chromosome 7q21 and mouse proximal chromosome 6 reveals a placental-specific imprinted gene, TFPI2/Tfpi2, which requires EHMT2 and EED for allelic-silencing David Monk,1,6 Alexandre Wagschal,2 Philippe Arnaud,2 Pari-Sima Mu¨ller,3 Layla Parker-Katiraee,4 Déborah Bourc’his,5 Stephen W. Scherer,4 Robert Feil,2 Philip Stanier,1 and Gudrun E. Moore1 1Institute of Child Health, London WC1N 1EH, United Kingdom; 2Institute of Molecular Genetics, CNRS UMR-5535 and University of Montpellier-II, 34293 Montpellier, France; 3Sir William Dunn School of Pathology, University of Oxford, Oxford OX1 3RE, United Kingdom; 4Center for Applied Genomics, The Hospital for Sick Children, Toronto M5G 1L7, Canada; 5Inserm U741, F-75251 Paris Cedex 05, France Genomic imprinting is a developmentally important mechanism that involves both differential DNA methylation and allelic histone modifications. Through detailed comparative characterization, a large imprinted domain mapping to chromosome 7q21 in humans and proximal chromosome 6 in mice was redefined. This domain is organized around a maternally methylated CpG island comprising the promoters of the adjacent PEG10 and SGCE imprinted genes. Examination of Dnmt3l−/+ conceptuses shows that imprinted expression for all genes of the cluster depends upon the germline methylation at this putative “imprinting control region” (ICR). Similarly as for other ICRs, we find its DNA-methylated allele to be associated with trimethylation of lysine 9 on histone H3 (H3K9me3) and trimethylation of lysine 20 on histone H4 (H4K20me3), whereas the transcriptionally active paternal allele is enriched in H3K4me2 and H3K9 acetylation. -

The Arf1p Gtpase-Activating Protein Glo3p Executes Its Regulatory Function Through a Conserved Repeat Motif at Its C-Terminus
2604 Research Article The Arf1p GTPase-activating protein Glo3p executes its regulatory function through a conserved repeat motif at its C-terminus Natsuko Yahara1,*, Ken Sato1,2 and Akihiko Nakano1,3,‡ 1Molecular Membrane Biology Laboratory, RIKEN Discovery Research Institute and 2PRESTO, Japan Science and Technology Agency, Hirosawa, Wako, Saitama 351-0198, Japan 3Department of Biological Sciences, Graduate School of Science, University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan *Present address: Department of Biochemistry, University of Geneva, Sciences II, Geneva, Switzerland ‡Author for correspondence (e-mail: [email protected]) Accepted 21 March 2006 Journal of Cell Science 119, 2604-2612 Published by The Company of Biologists 2006 doi:10.1242/jcs.02997 Summary ADP-ribosylation factors (Arfs), key regulators of ArfGAP, Gcs1p, we have shown that the non-catalytic C- intracellular membrane traffic, are known to exert multiple terminal region of Glo3p is required for the suppression of roles in vesicular transport. We previously isolated eight the growth defect in the arf1 ts mutants. Interestingly, temperature-sensitive (ts) mutants of the yeast ARF1 gene, Glo3p and its homologues from other eukaryotes harbor a which showed allele-specific defects in protein transport, well-conserved repeated ISSxxxFG sequence near the C- and classified them into three groups of intragenic terminus, which is not found in Gcs1p and its homologues. complementation. In this study, we show that the We name this region the Glo3 motif and present evidence overexpression of Glo3p, one of the GTPase-activating that the motif is required for the function of Glo3p in vivo. proteins of Arf1p (ArfGAP), suppresses the ts growth of a particular group of the arf1 mutants (arf1-16 and arf1-17). -
![Views [10-12] in Favour of Information Theory- Methods](https://docslib.b-cdn.net/cover/2277/views-10-12-in-favour-of-information-theory-methods-1372277.webp)
Views [10-12] in Favour of Information Theory- Methods
BMC Bioinformatics BioMed Central Research article Open Access Evaluation of GO-based functional similarity measures using S. cerevisiae protein interaction and expression profile data Tao Xu1,2, LinFang Du*2 and Yan Zhou*3,1 Address: 1Shanghai-MOST Key Laboratory of Health and Disease Genomics, Chinese National Human Genome Center at Shanghai, Shanghai 201203, PR China, 2College of Life Sciences, Sichuan University, Chengdu 610064, PR China and 3Department of Microbiology, School of Life Sciences, Fudan University, Shanghai 200433, PR China Email: [email protected]; LinFangDu*[email protected]; Yan Zhou* - [email protected] * Corresponding authors Published: 6 November 2008 Received: 18 March 2008 Accepted: 6 November 2008 BMC Bioinformatics 2008, 9:472 doi:10.1186/1471-2105-9-472 This article is available from: http://www.biomedcentral.com/1471-2105/9/472 © 2008 Xu et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Researchers interested in analysing the expression patterns of functionally related genes usually hope to improve the accuracy of their results beyond the boundaries of currently available experimental data. Gene ontology (GO) data provides a novel way to measure the functional relationship between gene products. Many approaches have been reported for calculating the similarities between two GO terms, known as semantic similarities. However, biologists are more interested in the relationship between gene products than in the scores linking the GO terms. -

Mouse Bet1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Bet1 Knockout Project (CRISPR/Cas9) Objective: To create a Bet1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Bet1 gene (NCBI Reference Sequence: NM_009748 ; Ensembl: ENSMUSG00000032757 ) is located on Mouse chromosome 6. 4 exons are identified, with the ATG start codon in exon 1 and the TGA stop codon in exon 4 (Transcript: ENSMUST00000049166). Exon 1~4 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 1 starts from about 0.28% of the coding region. Exon 1~4 covers 100.0% of the coding region. The size of effective KO region: ~8910 bp. The KO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 3 4 Legends Exon of mouse Bet1 Knockout region Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of start codon is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. The gRNA site is selected outside of these tandem repeats. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of stop codon is aligned with itself to determine if there are tandem repeats. -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Protein Tyrosine Phosphorylation in Haematopoietic Cancers and the Functional Significance of Phospho- Lyn SH2 Domain
Protein Tyrosine Phosphorylation in Haematopoietic Cancers and the Functional Significance of Phospho- Lyn SH2 Domain By Lily Li Jin A thesis submitted in conformity with the requirements for the degree of Ph.D. in Molecular Genetics, Graduate Department of Molecular Genetics, in the University of Toronto © Copyright by Lily Li Jin (2015) Protein Tyrosine Phosphorylation in Haematopoietic Cancers and the Functional Significance of Phospho-Lyn SH2 Domain Lily Li Jin 2015 Ph.D. in Molecular Genetics Graduate Department of Molecular Genetics University of Toronto Abstract Protein-tyrosine phosphorylation (pY) is a minor but important protein post-translational modification that modulates a wide range of cellular functions and is involved in cancer. Dysregulation of tyrosine kinases (TKs) and protein-tyrosine phosphatases (PTPs) have been observed in multiple myeloma (MM) and acute myeloid leukemia (AML) and is a subject of study. Using recently developed mass spectrometry-based proteomics techniques, quantitative PTP expression and cellular pY profiles were generated for MM cell lines and mouse xenograft tumors, as well as primary AML samples. Integrated comprehensive analyses on these data implicated a subset of TKs and PTPs in MM and AML, with valuable insights gained on the dynamic regulation of pY in biological systems. In particular, I propose a model that describes the cellular pY state as a functional output of the total activated TKs and PTPs in the cell. My results show that the global pY profile in the cancer models is quantitatively related to the cellular levels of activated TKs and PTPs. Furthermore, the identity of the implicated TK/PTPs is system- ii dependent, demonstrating context-dependent regulation of pY. -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV). -

UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Cardiac Stretch-Induced Transcriptomic Changes are Axis-Dependent Permalink https://escholarship.org/uc/item/7m04f0b0 Author Buchholz, Kyle Stephen Publication Date 2016 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Cardiac Stretch-Induced Transcriptomic Changes are Axis-Dependent A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioengineering by Kyle Stephen Buchholz Committee in Charge: Professor Jeffrey Omens, Chair Professor Andrew McCulloch, Co-Chair Professor Ju Chen Professor Karen Christman Professor Robert Ross Professor Alexander Zambon 2016 Copyright Kyle Stephen Buchholz, 2016 All rights reserved Signature Page The Dissertation of Kyle Stephen Buchholz is approved and it is acceptable in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California, San Diego 2016 iii Dedication To my beautiful wife, Rhia. iv Table of Contents Signature Page ................................................................................................................... iii Dedication .......................................................................................................................... iv Table of Contents ................................................................................................................ v List of Figures ................................................................................................................... -

Temporal Uncoupling of the DNA Methylome and Transcriptional Repression During Embryogenesis
Downloaded from genome.cshlp.org on February 11, 2016 - Published by Cold Spring Harbor Laboratory Press Research Temporal uncoupling of the DNA methylome and transcriptional repression during embryogenesis Ozren Bogdanovic´,1 Steven W. Long,2 Simon J. van Heeringen,1 Arie B. Brinkman,1 Jose Luis Go´mez-Skarmeta,3 Hendrik G. Stunnenberg,1 Peter L. Jones,2 and Gert Jan C. Veenstra1,4 1Department of Molecular Biology, Faculty of Science, Nijmegen Centre for Molecular Life Sciences, Radboud University Nijmegen, 6500 Nijmegen, The Netherlands; 2Department of Cell and Developmental Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, USA; 3Centro Andaluz de Biologı´a del Desarrollo, Consejo Superior de Investigaciones Cientı´ficas and Universidad Pablo de Olavide, Carretera de Utrera Km1, 41013 Sevilla, Spain DNA methylation is a tightly regulated epigenetic mark associated with transcriptional repression. Next-generation se- quencing of purified methylated DNA obtained from early Xenopus tropicalis embryos demonstrates that this genome is heavily methylated during blastula and gastrula stages. Although DNA methylation is largely absent from transcriptional start sites marked with histone H3 lysine 4 trimethylation (H3K4me3), we find both promoters and gene bodies of active genes robustly methylated. In contrast, DNA methylation is absent in large H3K27me3 domains, indicating that these two repression pathways have different roles. Comparison with chromatin state maps of human ES cells reveals strong con- servation of epigenetic makeup and gene regulation between the two systems. Strikingly, genes that are highly expressed in pluripotent cells and in Xenopus embryos but not in differentiated cells exhibit relatively high DNA methylation. Therefore, we tested the repressive potential of DNA methylation using transient and transgenic approaches and show that meth- ylated promoters are robustly transcribed in blastula- and gastrula-stage embryos, but not in oocytes or late embryos. -
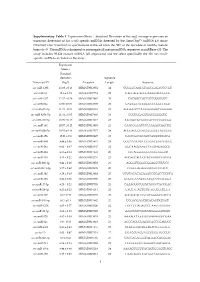
Supplementary Table 1. Expression
Supplementary Table 1. Expression (Mean Standard Deviation of the log2 average expression or transcript detection) of Sus scrofa specific miRNAs detected by the GeneChip™ miRNA 4.0 Array (ThermoFisher Scientific) in spermatozoa retrieved from the SRF of the ejaculate of healthy mature boars (n=3). The miRNA is designed to interrogate all mature miRNA sequences in miRBase v20. The array includes 30.424 mature miRNA (all organisms) and we select specifically the 326 Sus scrofa- specific miRNAs included in the array. Expression Mean ± Standard Deviation Sequence Transcript ID (log2) Accession Length Sequence ssc-miR-1285 13.98 ± 0.13 MIMAT0013954 24 CUGGGCAACAUAGCGAGACCCCGU ssc-miR-16 12.6 ± 0.74 MIMAT0007754 22 UAGCAGCACGUAAAUAUUGGCG ssc-miR-4332 12.32 ± 0.29 MIMAT0017962 20 CACGGCCGCCGCCGGGCGCC ssc-miR-92a 12.06 ± 0.09 MIMAT0013908 22 UAUUGCACUUGUCCCGGCCUGU ssc-miR-671-5p 11.73 ± 0.54 MIMAT0025381 24 AGGAAGCCCUGGAGGGGCUGGAGG ssc-miR-4334-5p 11.31 ± 0.05 MIMAT0017966 19 CCCUGGAGUGACGGGGGUG ssc-miR-425-5p 10.99 ± 0.15 MIMAT0013917 23 AAUGACACGAUCACUCCCGUUGA ssc-miR-191 10.57 ± 0.22 MIMAT0013876 23 CAACGGAAUCCCAAAAGCAGCUG ssc-miR-92b-5p 10.53 ± 0.18 MIMAT0017377 24 AGGGACGGGACGCGGUGCAGUGUU ssc-miR-15b 10.01 ± 0.9 MIMAT0002125 22 UAGCAGCACAUCAUGGUUUACA ssc-miR-30d 9.89 ± 0.36 MIMAT0013871 24 UGUAAACAUCCCCGACUGGAAGCU ssc-miR-26a 9.62 ± 0.47 MIMAT0002135 22 UUCAAGUAAUCCAGGAUAGGCU ssc-miR-484 9.55 ± 0.14 MIMAT0017974 20 CCCAGGGGGCGACCCAGGCU ssc-miR-103 9.53 ± 0.22 MIMAT0002154 23 AGCAGCAUUGUACAGGGCUAUGA ssc-miR-296-3p 9.41 ± 0.26 MIMAT0022958