Beta Hemoglobinopathies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of Methemoglobin and Carboxyhemoglobin in COVID-19: a Review
Journal of Clinical Medicine Review The Role of Methemoglobin and Carboxyhemoglobin in COVID-19: A Review Felix Scholkmann 1,2,*, Tanja Restin 2, Marco Ferrari 3 and Valentina Quaresima 3 1 Biomedical Optics Research Laboratory, Department of Neonatology, University Hospital Zurich, University of Zurich, 8091 Zurich, Switzerland 2 Newborn Research Zurich, Department of Neonatology, University Hospital Zurich, University of Zurich, 8091 Zurich, Switzerland; [email protected] 3 Department of Life, Health and Environmental Sciences, University of L’Aquila, 67100 L’Aquila, Italy; [email protected] (M.F.); [email protected] (V.Q.) * Correspondence: [email protected]; Tel.: +41-4-4255-9326 Abstract: Following the outbreak of a novel coronavirus (SARS-CoV-2) associated with pneumonia in China (Corona Virus Disease 2019, COVID-19) at the end of 2019, the world is currently facing a global pandemic of infections with SARS-CoV-2 and cases of COVID-19. Since severely ill patients often show elevated methemoglobin (MetHb) and carboxyhemoglobin (COHb) concentrations in their blood as a marker of disease severity, we aimed to summarize the currently available published study results (case reports and cross-sectional studies) on MetHb and COHb concentrations in the blood of COVID-19 patients. To this end, a systematic literature research was performed. For the case of MetHb, seven publications were identified (five case reports and two cross-sectional studies), and for the case of COHb, three studies were found (two cross-sectional studies and one case report). The findings reported in the publications show that an increase in MetHb and COHb can happen in COVID-19 patients, especially in critically ill ones, and that MetHb and COHb can increase to dangerously high levels during the course of the disease in some patients. -

Hereditary Spherocytosis: Clinical Features
Title Overview: Hereditary Hematological Disorders of red cell shape. Disorders Red cell Enzyme disorders Disorders of Hemoglobin Inherited bleeding disorders- platelet disorders, coagulation factor Anthea Greenway MBBS FRACP FRCPA Visiting Associate deficiencies Division of Pediatric Hematology-Oncology Duke University Health Service Inherited Thrombophilia Hereditary Disorders of red cell Disorders of red cell shape (cytoskeleton): cytoskeleton: • Mutations of 5 proteins connect cytoskeleton of red cell to red cell membrane • Hereditary Spherocytosis- sphere – Spectrin (composed of alpha, beta heterodimers) –Ankyrin • Hereditary Elliptocytosis-ellipse, elongated forms – Pallidin (band 4.2) – Band 4.1 (protein 4.1) • Hereditary Pyropoikilocytosis-bizarre red cell forms – Band 3 protein (the anion exchanger, AE1) – RhAG (the Rh-associated glycoprotein) Normal red blood cell- discoid, with membrane flexibility Hereditary Spherocytosis: Clinical features: • Most common hereditary hemolytic disorder (red cell • Neonatal jaundice- severe (phototherapy), +/- anaemia membrane) • Hemolytic anemia- moderate in 60-75% cases • Mutations of one of 5 genes (chromosome 8) for • Severe hemolytic anaemia in 5% (AR, parents ASx) cytoskeletal proteins, overall effect is spectrin • fatigue, jaundice, dark urine deficiency, severity dependant on spectrin deficiency • SplenomegalSplenomegaly • 200-300:million births, most common in Northern • Chronic complications- growth impairment, gallstones European countries • Often follows clinical course of affected -

Structure and Function of Leghemoglobins*
203 Structure and function of leghemoglobins* by M. BECANA**, J.F. MORAN, I. ITURBE-ORMAETXE, Y. GOGORCENA and P.R. ESCUREDO Departamento de Nutrición Vegetal, Estación Experimental de Aula Dei (C.S.I.C.), Apartado 202, 50080 Zaragoza Received: 31-10-1994 Key words: Free radicals, Iron, Leghemoglobins, Nitrogen fixation, Oxygen, Plant senescence, Root nodules. Abbreviation: Lb, leghemoglobin. ABSTRACT Becana, M., Moran, J.F., Iturbe-Ormaetxe, I., Gogorcena, Y. and Escuredo, P.R. 1995. Structure and function of leghemoglobins. An. Estac. Exp. Aula Dei (Zaragoza) 21(3): 203-208. Leghemoglobin (Lb) is a myoglobin-like protein of about 16 kDa, which occurs in legume root nodules at very high concentra - tions. Usually the heme moiety is synthesized by the bacteroids but mitochondria may provide also heme for Lb when bacteria are defective in heme production or perhaps when Lb is produced in uninfected cells of nodules. Lb plays an essential role in the nitro - gen fixation process, by providing oxygen to the bacteroids at a low, but constant, concentration, which allows for simultaneous bac - teroid respiration and nitrogenase activity. Lb must be in the reduced, ferrous state to carry oxygen. Several factors within the nodu - les are conducive for Lb oxidation to its ferric, inactive form. During these inactivation reactions free radicals are generated. Howe - ver, healthy nodules contain around 80% of ferrous Lb and 20% of oxyferrous Lb, but not ferric Lb, which indicates that mechanisms exist in the nodules to maintain Lb reduced; these are the enzyme ferric Lb reductase and free flavins. Lb degradation is a largely unk - nown process, but several intermediates with modified hemes,presumably by oxidative attack,have been encountered, including modi - fied Lbam, choleglobin, and biliverdin. -

Alpha Thalassemia Trait
Alpha Thalassemia Trait Alpha Thalassemia Trait Produced by St. Jude Children’s Research Hospital, Departments of Hematology, Patient Education, 1 and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to replace counseling by a trained health care professional or genetic counselor. Our aim is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specific treatment options should be discussed with your doctor. For general information on sickle cell disease and other blood disorders, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital Alpha thalassemia trait All red blood cells contain hemoglobin (HEE muh glow bin), which carries oxygen from your lungs to all parts of your body. Alpha thalassemia (thal uh SEE mee uh) trait is a condition that affects the amount of hemo- globin in the red blood cells. • Adult hemoglobin (hemoglobin A) is made of alpha and beta globins. • Normally, people have 4 genes for alpha globin with 2 genes on each chromosome (aa/aa). People with alpha thalassemia trait only have 2 genes for alpha globin, so their bodies make slightly less hemoglobin than normal. This trait was passed on from their parents, like hair color or eye color. A trait is different from a disease 2 Alpha thalassemia trait is not a disease. Normally, a trait will not make you sick. Parents who have alpha thalassemia trait can pass it on to their children. -

Methemoglobinemia and Ascorbate Deficiency in Hemoglobin E Β Thalassemia: Metabolic and Clinical Implications
From www.bloodjournal.org by guest on April 2, 2015. For personal use only. Plenary paper Methemoglobinemia and ascorbate deficiency in hemoglobin E  thalassemia: metabolic and clinical implications Angela Allen,1,2 Christopher Fisher,1 Anuja Premawardhena,3 Dayananda Bandara,4 Ashok Perera,4 Stephen Allen,2 Timothy St Pierre,5 Nancy Olivieri,6 and David Weatherall1 1MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom; 2College of Medicine, Swansea University, Swansea, United Kingdom; 3University of Kelaniya, Colombo, Sri Lanka; 4National Thalassaemia Centre, District Hospital, Kurunegala, Sri Lanka; 5School of Physics, University of Western Australia, Crawley, Australia; and 6Hemoglobinopathy Research, University Health Network, Toronto, ON During investigations of the phenotypic man hypoxia induction factor pathway is There was, in addition, a highly signifi- diversity of hemoglobin (Hb) E  thalasse- not totally dependent on ascorbate lev- cant correlation between methemoglobin mia, a patient was encountered with per- els. A follow-up study of 45 patients with levels, splenectomy, and factors that sistently high levels of methemoglobin HbE  thalassemia showed that methemo- modify the degree of globin-chain imbal- associated with a left-shift in the oxygen globin levels were significantly increased ance. Because methemoglobin levels are dissociation curve, profound ascorbate and that there was also a significant re- modified by several mechanisms and may deficiency, and clinical features of scurvy; duction in plasma ascorbate levels. Hap- play a role in both adaptation to anemia these abnormalities were corrected by toglobin levels were significantly re- and vascular damage, there is a strong treatment with vitamin C. -

Chain of Human Neutrophil Cytochrome B CHARLES A
Proc. Nati. Acad. Sci. USA Vol. 85, pp. 3319-3323, May 1988 Biochemistry Primary structure and unique expression of the 22-kilodalton light chain of human neutrophil cytochrome b CHARLES A. PARKOS*, MARY C. DINAUERt, LESLIE E. WALKER*, RODGER A. ALLEN*, ALGIRDAS J. JESAITIS*, AND STUART H. ORKINtt *Department of Immunology, Research Institute of the Scripps Clinic, La Jolla, CA 92037; tDivision of Hematology-Oncology, Children's Hospital, and Dana-Farber Cancer Institute, Department of Pediatrics, Harvard Medical School, Boston, MA 02115; and tHoward Hughes Medical Institute, Children's Hospital, Boston, MA 02115 Communicated by Harvey F. Lodish, January 14, 1988 ABSTRACT Cytochrome b comprising 91-kDa and 22- Cytochrome b purified from neutrophil membranes ap- kDa subunits is a critical component of the membrane-bound pears to be a heterodimer of a glycosylated 91-kDa heavy oxidase of phagocytes that generates superoxide. This impor- chain and a nonglycosylated 22-kDa light chain (10-12). The tant microbicidal system is impaired in inherited disorders 91-kDa subunit is encoded by a gene designated CGD, known as chronic granulomatous disease (CGD). Previously we residing at chromosomal position Xp2l, which originally was determined the sequence of the larger subunit from the cDNA identified on the basis of genetic linkage without reference to of the CGD gene, the X chromosome locus affected in "X- a specific protein product (8). Antisera generated to either a linked" CGD. To complete the primary structure of the synthetic peptide predicted from the cDNA or to a fusion cytochrome b and to assess expression of the smaller subunit, protein produced in E. -

Table S1. Identified Proteins with Exclusive Expression in Cerebellum of Rats of Control, 10Mg F/L and 50Mg F/L Groups
Table S1. Identified proteins with exclusive expression in cerebellum of rats of control, 10mg F/L and 50mg F/L groups. Accession PLGS Protein Name Group IDa Score Q3TXS7 26S proteasome non-ATPase regulatory subunit 1 435 Control Q9CQX8 28S ribosomal protein S36_ mitochondrial 197 Control P52760 2-iminobutanoate/2-iminopropanoate deaminase 315 Control Q60597 2-oxoglutarate dehydrogenase_ mitochondrial 67 Control P24815 3 beta-hydroxysteroid dehydrogenase/Delta 5-->4-isomerase type 1 84 Control Q99L13 3-hydroxyisobutyrate dehydrogenase_ mitochondrial 114 Control P61922 4-aminobutyrate aminotransferase_ mitochondrial 470 Control P10852 4F2 cell-surface antigen heavy chain 220 Control Q8K010 5-oxoprolinase 197 Control P47955 60S acidic ribosomal protein P1 190 Control P70266 6-phosphofructo-2-kinase/fructose-2_6-bisphosphatase 1 113 Control Q8QZT1 Acetyl-CoA acetyltransferase_ mitochondrial 402 Control Q9R0Y5 Adenylate kinase isoenzyme 1 623 Control Q80TS3 Adhesion G protein-coupled receptor L3 59 Control B7ZCC9 Adhesion G-protein coupled receptor G4 139 Control Q6P5E6 ADP-ribosylation factor-binding protein GGA2 45 Control E9Q394 A-kinase anchor protein 13 60 Control Q80Y20 Alkylated DNA repair protein alkB homolog 8 111 Control P07758 Alpha-1-antitrypsin 1-1 78 Control P22599 Alpha-1-antitrypsin 1-2 78 Control Q00896 Alpha-1-antitrypsin 1-3 78 Control Q00897 Alpha-1-antitrypsin 1-4 78 Control P57780 Alpha-actinin-4 58 Control Q9QYC0 Alpha-adducin 270 Control Q9DB05 Alpha-soluble NSF attachment protein 156 Control Q6PAM1 Alpha-taxilin 161 -

Hemoglobin Wayne Trait with Incidental Polycythemia
Available online at www.annclinlabsci.org 96 Annals of Clinical & Laboratory Science, vol. 47, no. 1, 2017 Hemoglobin Wayne Trait with Incidental Polycythemia Manju Ambelil, Nghia Nguyen, Amitava Dasgupta, Semyon Risin, and Amer Wahed Department of Pathology and Laboratory Medicine, University of Texas Health Science Center- McGovern Medical School, Houston, TX, USA Abstract. Hemoglobinopathies, caused by mutations in the globin genes, are one of the most common inherited disorders. Many of the hemoglobin variants can be identified by hemoglobin analysis using conventional electrophoresis and high performance liquid chromatography; however hemoglobin DNA analysis may be necessary in other cases for confirmation. Here, we report a case of a rare alpha chain he- moglobin variant, hemoglobin Wayne, in a 47-year-old man who presented with secondary polycythemia. Capillary zone electrophoresis and high performance liquid chromatography revealed a significant amount of a hemoglobin variant, which was further confirmed by hemoglobin DNA sequencing as hemoglobin Wayne. Since the patient was not homozygous for hemoglobin Wayne, which is associated with second- ary polycythemia, the laboratory diagnosis in this case was critical in ruling out hemoglobinopathy as the etiology of his polycythemia. Key words: Hemoglobinopathies; hemoglobin Wayne; alpha globin chain variant. Introduction mean corpuscular hemoglobin of 27.1 pg. The red cell distribution width, total and differential leukocyte Hemoglobinopathies are the most common and counts, and platelets were normal. An anemia study clinically significant single-gene-inherited disorders showed a serum iron level of 127 mcg/dL, a ferritin con- [1]. These are caused by mutations in the globin centration of 239 ng/mL, an iron saturation of 42%, a total iron binding capacity of 306 mcg/dL, an unsatu- genes, resulting in quantitative and qualitative de- rated iron binding capacity of 179mcg/dL, and a vita- fects in the globin chain synthesis [2]. -
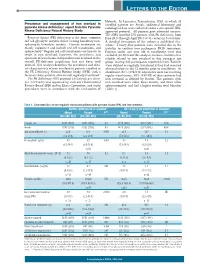
Prevalence and Management of Iron Overload in Pyruvate Kinase Deficiency
LETTERS TO THE EDITOR Helsinki. In Lancaster, Pennsylvania, USA, in which all Prevalence and management of iron overload in enrolled patients are Amish, additional laboratory and pyruvate kinase deficiency: report from the Pyruvate radiological data were collected under a site-specific IRB- Kinase Deficiency Natural History Study approved protocol. All patients gave informed consent. The NHS enrolled 278 patients with PK deficiency from Pyruvate kinase (PK) deficiency is the most common June 2014 through April 2017 at 31 centers in 6 countries. red cell glycolytic enzyme defect causing hereditary non- A detailed description of the cohort is published else- spherocytic hemolytic anemia. Current treatments are where.2 Twenty-four patients were excluded due to the mainly supportive and include red cell transfusions and inability to confirm two pathogenic PKLR mutations. splenectomy.1 Regular red cell transfusions are known to Patients under one year old at enrollment were also result in iron overload; however, the prevalence and excluded (n=12) from this analysis, because ferritin is less spectrum of transfusion-independent iron overload in the reliably related to iron overload in this youngest age overall PK-deficient population has not been well group, leaving 242 participants reported herein. Patients defined. This analysis describes the prevalence and clini- were defined as regularly transfused if they had received cal characteristics of iron overload in patients enrolled in ≥6 transfusions in the 12 months prior to enrollment. At the PK Deficiency Natural History Study (NHS) with a enrollment, 82% (198/242) of patients were not receiving focus on those patients who are not regularly transfused.2 regular transfusions; 38% (53/138) of these patients had The PK deficiency NHS protocol (clinicaltrials.gov identi- iron overload as defined by ferritin. -

Molecular Profiling of Innate Immune Response Mechanisms in Ventilator-Associated 2 Pneumonia
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.08.899294; this version posted January 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title: Molecular profiling of innate immune response mechanisms in ventilator-associated 2 pneumonia 3 Authors: Khyatiben V. Pathak1*, Marissa I. McGilvrey1*, Charles K. Hu3, Krystine Garcia- 4 Mansfield1, Karen Lewandoski2, Zahra Eftekhari4, Yate-Ching Yuan5, Frederic Zenhausern2,3,6, 5 Emmanuel Menashi3, Patrick Pirrotte1 6 *These authors contributed equally to this work 7 Affiliations: 8 1. Collaborative Center for Translational Mass Spectrometry, Translational Genomics Research 9 Institute, Phoenix, Arizona 85004 10 2. Translational Genomics Research Institute, Phoenix, Arizona 85004 11 3. HonorHealth Clinical Research Institute, Scottsdale, Arizona 85258 12 4. Applied AI and Data Science, City of Hope Medical Center, Duarte, California 91010 13 5. Center for Informatics, City of Hope Medical Center, Duarte, California 91010 14 6. Center for Applied NanoBioscience and Medicine, University of Arizona, Phoenix, Arizona 15 85004 16 Corresponding author: 17 Dr. Patrick Pirrotte 18 Collaborative Center for Translational Mass Spectrometry 19 Translational Genomics Research Institute 20 445 North 5th Street, Phoenix, AZ, 85004 21 Tel: 602-343-8454; 22 [[email protected]] 23 Running Title: Innate immune response in ventilator-associated pneumonia 24 Key words: ventilator-associated pneumonia; endotracheal aspirate; proteome, metabolome; 25 neutrophil degranulation 26 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.01.08.899294; this version posted January 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Identifying Genetic Variants and Pathways Associated with Extreme Levels of Fetal Hemoglobin in Sickle Cell Disease in Tanzania
Nkya et al. BMC Medical Genetics (2020) 21:125 https://doi.org/10.1186/s12881-020-01059-1 RESEARCH ARTICLE Open Access Identifying genetic variants and pathways associated with extreme levels of fetal hemoglobin in sickle cell disease in Tanzania Siana Nkya1,2*, Liberata Mwita2, Josephine Mgaya2, Happiness Kumburu3, Marco van Zwetselaar3, Stephan Menzel4, Gaston Kuzamunu Mazandu5,6,7* , Raphael Sangeda2,8, Emile Chimusa5 and Julie Makani2 Abstract Background: Sickle cell disease (SCD) is a blood disorder caused by a point mutation on the beta globin gene resulting in the synthesis of abnormal hemoglobin. Fetal hemoglobin (HbF) reduces disease severity, but the levels vary from one individual to another. Most research has focused on common genetic variants which differ across populations and hence do not fully account for HbF variation. Methods: We investigated rare and common genetic variants that influence HbF levels in 14 SCD patients to elucidate variants and pathways in SCD patients with extreme HbF levels (≥7.7% for high HbF) and (≤2.5% for low HbF) in Tanzania. We performed targeted next generation sequencing (Illumina_Miseq) covering exonic and other significant fetal hemoglobin-associated loci, including BCL11A, MYB, HOXA9, HBB, HBG1, HBG2, CHD4, KLF1, MBD3, ZBTB7A and PGLYRP1. Results: Results revealed a range of genetic variants, including bi-allelic and multi-allelic SNPs, frameshift insertions and deletions, some of which have functional importance. Notably, there were significantly more deletions in individuals with high HbF levels (11% vs 0.9%). We identified frameshift deletions in individuals with high HbF levels and frameshift insertions in individuals with low HbF. CHD4 and MBD3 genes, interacting in the same sub-network, were identified to have a significant number of pathogenic or non-synonymous mutations in individuals with low HbF levels, suggesting an important role of epigenetic pathways in the regulation of HbF synthesis. -

Sickle Cell Disease
Sickle cell disease Description Sickle cell disease is a group of disorders that affects hemoglobin, the molecule in red blood cells that delivers oxygen to cells throughout the body. People with this disease have atypical hemoglobin molecules called hemoglobin S, which can distort red blood cells into a sickle, or crescent, shape. Signs and symptoms of sickle cell disease usually begin in early childhood. Characteristic features of this disorder include a low number of red blood cells (anemia), repeated infections, and periodic episodes of pain. The severity of symptoms varies from person to person. Some people have mild symptoms, while others are frequently hospitalized for more serious complications. The signs and symptoms of sickle cell disease are caused by the sickling of red blood cells. When red blood cells sickle, they break down prematurely, which can lead to anemia. Anemia can cause shortness of breath, fatigue, and delayed growth and development in children. The rapid breakdown of red blood cells may also cause yellowing of the eyes and skin, which are signs of jaundice. Painful episodes can occur when sickled red blood cells, which are stiff and inflexible, get stuck in small blood vessels. These episodes deprive tissues and organs, such as the lungs, kidneys, spleen, and brain, of oxygen-rich blood and can lead to organ damage. A particularly serious complication of sickle cell disease is high blood pressure in the blood vessels that supply the lungs (pulmonary hypertension), which can lead to heart failure. Pulmonary hypertension occurs in about 10 percent of adults with sickle cell disease. Frequency Sickle cell disease affects millions of people worldwide.