162998571.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Global Insecticide Use for Vector-Borne Disease Control
WHO/CDS/NTD/WHOPES/GCDPP/2007.2 GLOBAL INSECTICIDE USE FOR VECTOR-BORNE DISEASE CONTROL M. Zaim & P. Jambulingam DEPARTMENT OF CONTROL OF NEGLECTED TROPICAL DISEASES (NTD) WHO PESTICIDE EVALUATION SCHEME (WHOPES) First edition, 2002 Second edition, 2004 Third edition, 2007 © World Health Organization 2007 All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. All reasonable precautions have been taken by the World Health Organization to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either express or implied. The responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health Organization be liable for damages arising from its use. The named authors alone are responsible for the views expressed in this publication. CONTENTS Page Acknowledgements i Introduction 1 Collection of information 2 Data analysis and observations on reporting 3 All uses in vector control 6 Malaria vector control 22 Dengue vector control 38 Chagas disease vector control 48 Leishmaniasis vector control 52 Other vector-borne disease control 56 Selected insecticides – DDT 58 Selected insecticides – Insect growth regulators 60 Selected insecticides – Bacterial larvicides 62 Country examples 64 Annex 1. -

Full Page Fax Print
RESISTANCE TO MALATHION, PIRIMIPHOS-METHYL AND FENITROTHION IN COLEOPTERA FROM STORED GRAINS. Ivania A. PACHECO; M. Regina SARTORI; Scheilla BOLONHEZI Instituto de Tecnologia de Alimentos Avenida Brasil, 2880 - Caixa Postal 139 Campinas - Sao Paulo - Brasil - CEP 13073 SUMMARY The objectives of this paper were:l) to verify the occurrance of resistance to malathion, pirimiphos-methyl and fenitrothion in populations of coleoptera from stored grains and 2) to obtain data that could contribute to the adequate control of these insects during storage, thus reducing losses. Populations of Sitophilus oryzae, ~. zeamais, Tribolium castaneum and Rhyzopertha dominica were collected from storage facilities located in different regions of Brazil, and tested for resistance to the tree insecticides according to the FAO Standard Method (FAO, 1975). Insects were collected in the States of Sao Paulo, Rio Grande do SuI, Santa Catarina, Goias, Acre and Rondonia. Twenty populations of S. oryzae, ten of S. zeamais, twenty of R. dominica and twenty -five of T. castan~ad already been tested for - resistance to the three insecticides. Seven populations of S. oryzae, six of R. dominica and ten of S. zean~is were susceptible 'to the th~ee insecticides. Resistance to malathion was showed in thirteen populations of S. oryzae, fourteen of ~. dominica and twenty-five of !. castaneum. Simultaneous cross-resistance to pirimiphos-methyl and fenitrothion was indicated in one population of ~. oryzae and eight of !. castaneum. Cross resistance only to pirimiphos-methyl was indicated in two populations of ~. oryzae, one of ~. dominica and one of !. castaneum and only to fenitrothion in two of R. dominica and two of T. -

Pesticides and Toxic Substances
UNITED STATES ENVIRONMENTAL PROTECTION AGENCY WASHINGTON D.C., 20460 OFFICE OF PREVENTION, PESTICIDES AND TOXIC SUBSTANCES MEMORANDUM DATE: July 31, 2006 SUBJECT: Finalization of Interim Reregistration Eligibility Decisions (IREDs) and Interim Tolerance Reassessment and Risk Management Decisions (TREDs) for the Organophosphate Pesticides, and Completion of the Tolerance Reassessment and Reregistration Eligibility Process for the Organophosphate Pesticides FROM: Debra Edwards, Director Special Review and Reregistration Division Office of Pesticide Programs TO: Jim Jones, Director Office of Pesticide Programs As you know, EPA has completed its assessment of the cumulative risks from the organophosphate (OP) class of pesticides as required by the Food Quality Protection Act of 1996. In addition, the individual OPs have also been subject to review through the individual- chemical review process. The Agency’s review of individual OPs has resulted in the issuance of Interim Reregistration Eligibility Decisions (IREDs) for 22 OPs, interim Tolerance Reassessment and Risk Management Decisions (TREDs) for 8 OPs, and a Reregistration Eligibility Decision (RED) for one OP, malathion.1 These 31 OPs are listed in Appendix A. EPA has concluded, after completing its assessment of the cumulative risks associated with exposures to all of the OPs, that: (1) the pesticides covered by the IREDs that were pending the results of the OP cumulative assessment (listed in Attachment A) are indeed eligible for reregistration; and 1 Malathion is included in the OP cumulative assessment. However, the Agency has issued a RED for malathion, rather than an IRED, because the decision was signed on the same day as the completion of the OP cumulative assessment. -

Qsar Analysis of the Chemical Hydrolysis of Organophosphorus Pesticides in Natural Waters
QSAR ANALYSIS OF THE CHEMICAL HYDROLYSIS OF ORGANOPHOSPHORUS PESTICIDES IN NATURAL WATERS. by Kenneth K. Tanji Principal Investigator and Jonathan 1. Sullivan Graduate Research Assistant Department of Land, Air and Water Resources University of California, Davis Technical Completion Report Project Number W-843 August, 1995 University of California Water Resource Center The research leading to this report was supported by the University of California Water Resource Center as part of Water Resource Center Project W-843. Table of Contents Page Abstract 2 Problem and Research Objectives 3 Introduction 5 Theoretical Background 6 QSAR Methodology 7 Molecular Connectivity Theory 8 Organophosphorus Pesticides 12 Experimental Determination of Rates 15 Results and Discussion 17 Principal Findings and Significance 19 References 34 List of Tables Page Table 1. Statistical relationship between OP pesticides and first-order MC/'s. 30 Table 2. Inherent conditions of waters used in experimental work. 16 Table 3. Estimated half-lives for organophosphorus esters derived from model. 31 Table 4. Half-lives and first-order MCI' sfor model calibration data set. 31 Table 5. Experimental kinetic data for validation set compounds, Sacramento. 33 List of Figures Page Figure 1. Essential Features OfQSAR Modeling Methodology. 21 Figure 2. Regression plot for In hydrolysis rate vs. 1st order MCl' s. 22 Figure 3. a 3-D molecular model, a line-segment model and a graphical model. 23 Figure 4. Molecular connectivity index suborders. 24 Figure 5. Chlorpyrifos and its fourteen fourth order path/cluster fragments. 25 Figure 6. Abridged MClndex output. 26 Figure 7. Parent acids of most common organophosphorus pesticides. 12 Figure 8. -

Environmental Health Criteria 63 ORGANOPHOSPHORUS
Environmental Health Criteria 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION Please note that the layout and pagination of this web version are not identical with the printed version. Organophophorus insecticides: a general introduction (EHC 63, 1986) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1986 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination -
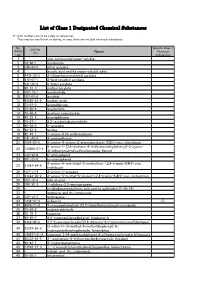
List of Class 1 Designated Chemical Substances
List of Class 1 Designated Chemical Substances *1:CAS numbers are to be solely as references. They may be insufficient or lacking, in case there are multiple chemical substances. No. Specific Class 1 CAS No. (PRTR Chemical (*1) Name Law) Substances 1 - zinc compounds(water-soluble) 2 79-06-1 acrylamide 3 140-88-5 ethyl acrylate 4 - acrylic acid and its water-soluble salts 5 2439-35-2 2-(dimethylamino)ethyl acrylate 6 818-61-1 2-hydroxyethyl acrylate 7 141-32-2 n-butyl acrylate 8 96-33-3 methyl acrylate 9 107-13-1 acrylonitrile 10 107-02-8 acrolein 11 26628-22-8 sodium azide 12 75-07-0 acetaldehyde 13 75-05-8 acetonitrile 14 75-86-5 acetone cyanohydrin 15 83-32-9 acenaphthene 16 78-67-1 2,2'-azobisisobutyronitrile 17 90-04-0 o-anisidine 18 62-53-3 aniline 19 82-45-1 1-amino-9,10-anthraquinone 20 141-43-5 2-aminoethanol 21 1698-60-8 5-amino-4-chloro-2-phenylpyridazin-3(2H)-one; chloridazon 5-amino-1-[2,6-dichloro-4-(trifluoromethyl)phenyl]-3-cyano- 22 120068-37-3 4[(trifluoromethyl)sulfinyl]pyrazole; fipronil 23 123-30-8 p-aminophenol 24 591-27-5 m-aminophenol 4-amino-6-tert-butyl-3-methylthio-1,2,4-triazin-5(4H)-one; 25 21087-64-9 metribuzin 26 107-11-9 3-amino-1-propene 27 41394-05-2 4-amino-3-methyl-6-phenyl-1,2,4-triazin-5(4H)-one; metamitron 28 107-18-6 allyl alcohol 29 106-92-3 1-allyloxy-2,3-epoxypropane 30 - n-alkylbenzenesulfonic acid and its salts(alkyl C=10-14) 31 - antimony and its compounds 32 120-12-7 anthracene 33 1332-21-4 asbestos ○ 34 4098-71-9 3-isocyanatomethyl-3,5,5-trimethylcyclohexyl isocyanate 35 78-84-2 isobutyraldehyde -

Pesticide Resistance of Agricultural Pests in Japan
Pesticide Resistance of Agricultural Pests in Japan By TOSHIKAZU IWATA Chief, 3rd Laboratory of Insect Control Investigation, Department of Phytopathology and Entomology, National Institute of Agricultural Sciences A quarter century has passed since DDT other organophosphorus chemicals such as was firstly applied to control agricultural in methyl paration. As the result the parathion sect pests. The amount of synthesized organic resistant borer was prevented from spreading insecticides applied in fields increased rapidly by changing insecticide to be applied for the since 1952 and the rate of increase was about control. 10 per cent a year. In 1960 an evident report Control of the rice stem borer by BHC dust was made on insecticide resistance of insects, was established in about 1950, and application so 10 years have passed following the first of fine dust or granular formulation of BHC appearance of insecticide-resistant insects. to paddy fields became very popular in 1961 Today, pest species developed or suspected of or 1962. In 1964, declination of susceptibility resistance totals 11 species of insects and 6 of the borers to BHC was found in Kagawa species of mites as shown in Table 1. The Prefecture. After that, similar phenomena author reviews the circumstances of occur were reported in some other prefectures. The rence and some results of studies on major amount of BHC applied to paddy fields has instances of resistant pests. been decreased for its residue in rice plants. Consequently, BHC resistance of rice stem Rice stem borer, Chilo suppressalis borer may not become a serious problem in Walker future. Recently, declination of susceptibility of The rice stem borer is one of the most im the borer to fenitrothion is suspected in a portant pests of rice plants in Japan. -

Appendix Common, Trade, and Chemical Names of Pesticides Mentioned in the Present Volume
Appendix Common, trade, and chemical names of pesticides mentioned in the present volume Commonname Tradename Chemical name aldrin Octalene 1,2,3,4,10,1 O-hexachloro-1 ,4,4a,S,8,8a hexahydro-1,4-endo-exo-S,8-dimethano naphthalene amidithion Thiocron O,O-dimethyl S-(2-methoxyethyl carbamoylmethyl) phosphorodithioate azinphos methyl Guthion, Gusathion O,O-dimcthyl S-( 4-oxo-l ,2,3-benzotri azin-3-( 4H)-ylmethyl) phosphorodithioate captan Orthocid N -(trichloromethylthio) cyclohex-4-ene- 1,2-dicarboximide carbaryl Sevin 1-naphthyl-mcthylcarbamate carbophenothion Trithion O,O-diethyl S-[(p-chlorophenylthio) methyl] phosphorodithioate chlorfenvinphos Birlane (Shell), 2-chloro-1-(2,4-dichlorophenyl) vinyl Sapecron (e/BA) diethyl phosphate chlorphenamidine Galecron N'-( 4-chloro-o-tolyl)-N,N-dimethyl formamidine chlorthion Chlorthion O,O-dimethyIO-(3-chloro-4-nitro phenyl)-phosphorothioate coumaphos Asuntol, Co-Ral O,O-diethyl O-(3-chloro-4-methyl- 2-oxo-2H-1-benzopyran-7-yl) phosphorothioate DDT Gesarol 1,1,1-trichloro-2,2-bis(p-chlorophenyl) ethane demeton Systox O,O-diethyl O-(and S)-2-(ethylthio)ethyl phosphorothioates DEF S,S,S-tributyltrithiophosphate demeton methyl Metasystox O,O-dimethyl O-(and S)-2-(ethylthio) ethyl phosphorothioates diazinon Diazinon, Basudin O,O-diethyIO-(2-isopropyl-4-methyl- 6-pyrimidyl) phosphorothioate dichlorvos Vapona (Shell), O,O-dimethyl-2,2-dichlorovinyl Nuvan (e/BA) phosphate dicrotophos Bidrin (Shell), O,O-dimethyl O-(2-dimethyl-carbamyl-1- Carbicron (e/BA) methyl) vinyl phosphate dieldrin Octalox -

NMP-Free Formulations of Neonicotinoids
(19) & (11) EP 2 266 400 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 29.12.2010 Bulletin 2010/52 A01N 43/40 (2006.01) A01N 43/86 (2006.01) A01N 47/40 (2006.01) A01N 51/00 (2006.01) (2006.01) (2006.01) (21) Application number: 09305544.0 A01P 7/00 A01N 25/02 (22) Date of filing: 15.06.2009 (84) Designated Contracting States: (72) Inventors: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR • Gasse, Jean-Jacques HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL 27600 Saint-Aubin-Sur-Gaillon (FR) PT RO SE SI SK TR • Duchamp, Guillaume Designated Extension States: 92230 Gennevilliers (FR) AL BA RS • Cantero, Maria 92230 Gennevilliers (FR) (71) Applicant: NUFARM 92233 Gennevelliers (FR) (74) Representative: Cabinet Plasseraud 52, rue de la Victoire 75440 Paris Cedex 09 (FR) (54) NMP-free formulations of neonicotinoids (57) The invention relates to NMP-free liquid formulation comprising at least one nicotinoid and at least one aprotic polar component selected from the group comprising the compounds of formula I, II or III below, and mixtures thereof, wherein R1 and R2 independently represent H or an alkyl group having less than 5 carbons, preferably a methyl group, and n represents an integer ranging from 0 to 5, and to their applications. EP 2 266 400 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 266 400 A1 Description Technical Field of the invention 5 [0001] The invention relates to novel liquid formulations of neonicotinoids and to their use for treating plants, for protecting plants from pests and/or for controlling pests infestation. -

Interaction Profile for Mixture of Insecticides: Pyrethroids
44 5. References Abdel-Rahman A, Dechkovskaia AM, Goldstein LB, et al. 2004. Neurological deficits induced by malathion, DEET, and permethrin, alone or in combination in adult rats. J Toxicol Environ Health A 67(4):331-356. ATSDR. 1992. Public health assessment guidance manual. Atlanta, GA: U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry. ATSDR. 2003a. Toxicological profile for pyrethrins and pyrethroids. Agency for Toxic Substances and Disease Registry. http://www.atsdr.cdc.gov/ToxProfiles/tp155.pdf. March 29, 2013. ATSDR. 2004a. Guidance manual for the assessment of joint toxic action of chemical mixtures. Atlanta, GA: Agency for Toxic Substances and Disease Registry. http://www.atsdr.cdc.gov/interactionprofiles/IP-ga/ipga.pdf. April 2, 2013. ATSDR. 2004b. Interaction profile for benzene, toluene, ethylbenzene, and xylenes (BTEX). Agency for Toxic Substances and Disease Registry. http://www.atsdr.cdc.gov/interactionprofiles/IP- btex/ip05.pdf. March 29, 2013. Belden JB, Lydy MJ. 2006. Joint toxicity of chlorpyrifos and esfenvalerate to fathead minnows and midge larvae. Environ Toxicol Chem 25(2):623-629. Bosgra S, van Eijkeren JC, van der Schans MJ, et al. 2009. Toxicodynamic analysis of the inhibition of isolated human acetylcholinesterase by combinations of methamidophos and methomyl in vitro. Toxicol Appl Pharmacol 236(1):1-8. Burr SA, Ray DE. 2004. Structure-activity and interaction effects of 14 different pyrethroids on voltage- gated chloride ion channels. Toxicol Sci 77(2):341-346. Cao Z, Shafer TJ, Crofton KM, et al. 2011. Additivity of pyrethroid actions on sodium influx in cerebrocortical neurons in primary culture. -

Registration Division Conventional Pesticides -Branch and Product
Registration Division Conventional Pesticides - Branch and Product Manager (PM) Assignments For a list of Branch contacts, please click the following link: http://www2.epa.gov/pesticide-contacts/contacts-office-pesticide-programs-registration-division Branch FB=Fungicide Branch. FHB=Fungicide Herbicide Branch. HB=Herbicide Branch. Abbreviations: IVB*= Invertebrate-Vertebrate Branch 1, 2 or 3. MUERB=Minor Use and Emergency Response Branch. Chemical Branch PM 1-Decanol FHB RM 20 1-Naphthaleneacetamide FHB RM 20 2, 4-D, Choline salt HB RM 23 2,4-D HB RM 23 2,4-D, 2-ethylhexyl ester HB RM 23 2,4-D, butoxyethyl ester HB RM 23 2,4-D, diethanolamine salt HB RM 23 2,4-D, dimethylamine salt HB RM 23 2,4-D, isopropyl ester HB RM 23 2,4-D, isopropylamine salt HB RM 23 2,4-D, sodium salt HB RM 23 2,4-D, triisopropanolamine salt HB RM 23 2,4-DB HB RM 23 2,4-DP HB RM 23 2,4-DP, diethanolamine salt HB RM 23 2,4-DP-p HB RM 23 2,4-DP-p, 2-ethylhexyl ester FB RM 21 2,4-DP-p, DMA salt HB RM 23 2-EEEBC FB RM 21 2-Phenylethyl propionate FHB RM 20 4-Aminopyridine IVB3 RM 07 4-Chlorophenoxyacetic acid FB RM 22 4-vinylcyclohexene diepoxide IVB3 RM 07 Abamectin IVB3 RM 07 Acephate IVB2 RM 10 Acequinocyl IVB3 RM 01 Acetaminophen IVB3 RM 07 Acetamiprid IVB3 RM 01 Acetic acid, (2,4-dichlorophenoxy)-, compd. with methanamine (1:1) HB RM 23 Acetic acid, trifluoro- FHB RM 20 Acetochlor HB RM 25 Acibenzolar-s-methyl FHB RM 24 Acid Blue 9 HB RM 23 Acid Yellow 23 HB RM 23 Sunday, June 06, 2021 Page 1 of 17 Chemical Branch PM Acifluorfen HB RM 23 Acrinathrin IVB1 RM 03 -

Toxicological Evaluation of Parathion and Azinphosmethyl in Freshwater Model Ecosystems
Y\v\&*io 6>\o C Toxicological evaluation of parathion and azinphosmethyl in freshwater model ecosystems NN08201.81B :R.J.Dorîland Toxicological evaluationo fparathio nan d azinphosmethyl infreshwate rmode l ecosystems Promotor: dr.J.H . Koeman,hoogleraa r ind e toxicologie. R.J. Dortland Toxicological evaluation of parathion and azinphosmethyl in freshwater model ecosystems Proefschrift terverkrijgin gva nd egraa dva n doctori nd elandbouwwetenschappen , op gezagva nd erecto rmagnificus , dr.H.C .va nde rPlas , hoogleraari nd eorganisch e scheikunde, inhe topenbaa rt everdedige n opvrijda g2 0jun i198 0 desnamiddag st evie ruu ri nd eaul a vand eLandbouwhogeschoo l teWageninge n Centre for Agricultural Publishing and Documentation Wageningen - 1980 Astract Dortland,R.J. ,1980 .Toxicologica l evaluationo fparathio nan dazinphosmethy li n freshwatermode l ecosystems.Agric .Res .Rep .(Versl .landbouwk .Onderz. )898 , ISBN9 022 0073 24 ,(viii )+ 112p. ,3 3figs ,4 2tables ,13 1refs ,appendices , Eng.an dDutc hsummaries . Also:Doctora l thesis,Wageningen . Astud ywa smad eo fth epossibl ehazard so f long-termexposur e offreshwate recosys temst olo w (<1 m gm~3 )concentration s oforganophosphoru s insecticides.Range-fin ding,acut ean dsub-acut e (3weeks )laborator y toxicity trialswer ecarrie d outwit h parathion,azinphosmethyl ,diazinon ,malathio nan dparathion-methyl .Th e rateso f dissipation ofthes ecompound s from thewate r phaseo faquari awit han dwithou tplant s andsediment swer estudied .Th emai ntes tspecie swer eDaphni amagna ,Asellu