Genomic Sequencing of the Aquatic Fusarium Spp. QHM and BWC1 and Their
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2017 Annual Report 1 Definitions
* Bank of Jinzhou Co., Ltd. is not an authorized institution within the meaning of the Banking Ordinane (Chapter 155 of the Laws of Hong Kong), not subject to the supervision of the Hong Kong Monetary Authority, and not authorized to carry on banking and/or deposit-taking business in Hong Kong. Contents 2 Definitions 4 Chapter 1 Company Profile 7 Chapter 2 Financial Highlights 10 Chapter 3 Chairman ’s Statement 12 Chapter 4 President’s Statement 14 Chapter 5 Management Discussion and Analysis 71 Chapter 6 Changes in Ordinary Shares and Particulars of Shareholders 77 Chapter 7 Particulars of Preference Shares 79 Chapter 8 Directors, Supervisors, Senior Management, Employees and Organizations 98 Chapter 9 Corporate Governance Report 119 Chapter 10 Directors’ Report 127 Chapter 11 Supervisors’ Report 130 Chapter 12 Social Responsibility Report 132 Chapter 13 Internal Control and Internal Audit 136 Chapter 14 Important Events 139 Chapter 15 Independent Auditor’s Report 149 Chapter 16 Financial Statements 269 Chapter 17 Unaudited Supplementary Financial Information Bank of Jinzhou Co., Ltd. 2017 Annual Report 1 Definitions In this annual report, unless the context otherwise requires, the following terms shall have the meanings set out below: “A Share Offering” the Bank’s proposed initial public offering of not more than 1,927,000,000 A shares, which has been approved by the Shareholders on 29 June 2016 “Articles of Association” the articles of association of the Bank, as the same may be amended from time to time “the Bank”, “Bank of Jinzhou” -

Virtual Simulation Analysis of Rigid-Flexible Coupling Dynamics of Shearer with Clearance
Hindawi Shock and Vibration Volume 2018, Article ID 6179054, 18 pages https://doi.org/10.1155/2018/6179054 Research Article Virtual Simulation Analysis of Rigid-Flexible Coupling Dynamics of Shearer with Clearance Hongyue Chen,1,2 Kun Zhang ,1 Mingbo Piao,1 Xin Wang ,1 Jun Mao ,1,2 and Qiushuang Song3 1 School of Mechanical Engineering, Liaoning Technical University, No. 88, Yulong Road, Xihe District, Fuxin City, Liaoning Province 123000, China 2China National Coal Association, Dynamic Research for High-End Complete Integrated Coal Mining Equipment and Big Data Analysis Center, No. 88, Yulong Road, Xihe District, Fuxin City, Liaoning Province 123000, China 3ChinaCoalEnergyCompanyLimited(ChinaCoalEnergy),No.1,HuangsiStreet,ChaoyangDistrict,BeijingCity100120,China Correspondence should be addressed to Kun Zhang; [email protected] Received 30 October 2017; Revised 4 February 2018; Accepted 20 February 2018; Published 4 April 2018 Academic Editor: Mario Terzo Copyright © 2018 Hongyue Chen et al. Tis is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. A model for virtual simulation analysis of the rigid-fexible coupling of a shearer has been developed with the objective of addressing problems associated with lifetime mismatch and low reliability of pin rows of a scraper conveyor and the corresponding support mechanism of a shearer. Simulations were performed using the experimental roller load as stimulus. Results of the analysis demonstrate that the vertical cutting force on the roller serves to reduce the load on the plane support plates during shearer cutting, and the force on the right plane support plate is considerably smaller compared to that on the lef plane support plate along the direction of motion of the shearer. -
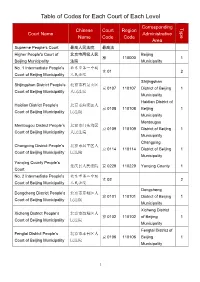
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

Geochemical Baseline Determination and Contamination of Heavy Metals in the Urban Topsoil of Fuxin City, China
ChinaXiv合作期刊 Geochemical baseline determination and contamination of heavy metals in the urban topsoil of Fuxin City, China ZHANG Hua, YU Miao, XU Hongjia, WEN Huan, FAN Haiyan, WANG Tianyi, LIU Jiangang* School of Geography, Liaoning Normal University, Dalian 116029, China Abstract: Urban topsoil is the most frequent interface between human society and natural environment. The accumulation of heavy metals in the urban topsoil has a direct effect on residents' life and health. The geochemical baseline of heavy metals is an objective description of the general level of heavy metals in the urban topsoil. Meanwhile, the determination of geochemical baseline is necessary for regional environmental management, especially in coal cities prone to heavy metal pollution. Heavy metal pollution has become an environmental problem in Fuxin City, China for a long time. To establish the geochemical baseline of heavy metals in the topsoil of Fuxin City and to evaluate the ecological risk of the topsoil, we collected 75 topsoil samples (0–20 cm) and analyzed the concentrations of Cu, Ni, Zn, Pb, Cr, Cd, Hg and As through X-ray fluorescence spectrometry, atomic absorption spectrometry and inductively coupled plasma optical emission spectrometry. We determined the geochemical baseline of heavy metals in the topsoil of Fuxin City by using iteration removal, box-whisker plot, cumulative frequency curve and reference metal normalization; evaluated the contamination risk and ecological risk of the topsoil by using the baseline factor index, Nemerow index and Hakanson potential ecological risk index; and identified the source category of heavy metals in the topsoil by using a pedigree clustering heatmap. Results showed that the geochemical baseline values were 42.86, 89.34, 92.23, 60.55, 145.21, 0.09, 0.08 and 4.17 mg/kg for Cu, Ni, Zn, Pb, Cr, Cd, Hg and As, respectively. -

Liaoning Safe and Sustainable Urban Water Supply Project
Document of The World Bank FOR OFFICIAL USE ONLY Public Disclosure Authorized Report No: PAD2096 INTERNATIONAL BANK FOR RECONSTRUCTION AND DEVELOPMENT PROJECT APPRAISAL DOCUMENT ON A PROPOSED LOAN Public Disclosure Authorized IN THE AMOUNT OF US$250 MILLION TO THE PEOPLE’S REPUBLIC OF CHINA FOR A LIAONING SAFE AND SUSTAINABLE URBAN WATER SUPPLY PROJECT Public Disclosure Authorized May 15, 2018 Water Global Practice East Asia and Pacific Region This document has a restricted distribution and may be used by recipients only in the performance of their official duties. Its contents may not otherwise be disclosed without World Bank authorization. Public Disclosure Authorized CURRENCY EQUIVALENTS (Exchange Rate Effective January 1, 2018) Currency Unit = Renminbi (RMB) Yuan (Y) RMB 1.00 = US$0.15 US$1.00 = RMB 6.48 FISCAL YEAR January 1 – December 31 ABBREVIATIONS AND ACRONYMS ADM Accountability and Decision-Making Framework (ADM) AWSC Anshan Water Supply Company BP Bank Procedure BPS Booster Pump Station CLG City Leading Group CNAO China National Auditing Office CO2 Carbon Dioxide CPMO City Project Management Office CPS Country Partnership Strategy CQS Consultant Qualifications based selection DA Designated Account DFIL Disbursement and Financial Information Letter DRC Development and Reform Commission EA Environmental Assessment EIRR Economic Internal Rate of Return EMP Environmental Management Plan EPB Environmental Protection Bureaus ESMAP Energy Sector Management Assistance Program ESMF Environmental and Social Management Framework FBS -

Current List. RECORD of APPROVED ACTIVE CONSTITUENTS for CHEMICAL PRODUCTS
CHEMICAL COMPANY MANUFACTURER SITE APPROVAL NO Current List RECORD OF APPROVED ACTIVE CONSTITUENTS FOR CHEMICAL PRODUCTS The Current Approval List is also accessible via the NRA web page on: http://www.nra.gov.au/tgac/tgac.pdf as at 10 December 2001 Page 1 of 152 CHEMICAL COMPANY MANUFACTURER SITE APPROVAL NO 1,3-dichloropropene Dow AgroSciences Australia Limited The Dow Chemical Company 52481 Building A-915 Freeport Texas 77541 USA 1,3-dichloropropene Dow AgroSciences Australia Limited Dow Chemical G.m.b.H. 52747 Werk Stade D-2160 Stade GERMANY 2-(Thiocyanomethylthio)benzothiazole Buckman Laboratories Ltd Buckman Laboratories Inc 44403 (TCMTB) 1256 North McLean Boulevard Memphis Tennessee 38108-0305 USA 2,4-D A H Marks Australia Limited A H Marks & Company Limited 51006 Wyke Lane Wyke Bradford West Yorkshire BD12 9EJ United Kingdom 2,4-D Artfern Pty Ltd Dalian Songliao Chemical Industrial Company Ltd 44245 22 Gongxing Road Ganjingzi Dalian Liaoning 116031 CHINA 2,4-D Ancom Australia Pty Ltd Ancom Crop Care SDN BHD 47430 Persiaran Selangor 40000 Shah Alam Selangor D.E. MALAYSIA Page 2 of 152 CHEMICAL COMPANY MANUFACTURER SITE APPROVAL NO 2,4-D Dow AgroSciences Australia Limited Sanachem (Pty) Ltd 47158 Hytor Road Chloorkop Kempton Park 1620 SOUTH AFRICA 2,4-D Dow AgroSciences Australia Limited Dow Agro Sciences (NZ) Ltd 47548 DowElanco (NZ) Limited Plant 1 89 Paritutu Road New Plymouth 4620 NEW ZEALAND 2,4-D Dow AgroSciences Australia Limited Dow Agro Sciences LLC 47550 The Dow Chemical Company Plant 1 Midland Michigan 48640 USA 2,4-D Farmoz Pty Ltd Choseright Limited 48691 Jin Jiang Agricultural Chemical Factory No 14 Hong Qiao North Road Jingjiang City Jiangsu Province CHINA 2,4-D Farmoz Pty Ltd Atanor S.A. -

Resettlement Action Plan
SFG3240 REV Public Disclosure Authorized World Bank Financed Liaoning Safe and Sustainable Urban Water Supply Project Public Disclosure Authorized Resettlement Action Plan Public Disclosure Authorized Public Disclosure Authorized Liaoning Province Urban and Rural Construction and Planning Design Institute May 2017 World Bank Financed Liaoning Safe and Sustainable Urban Water Supply Project Resettlement Action Plan President: Dong Yang General Engineer: Wang Guoqing Director of Municipal Zhao Hui Comprehensive Department: Person in Charge: Wang Xianming Team Member : Liu Yimin Chen Chao Dong Hongwei Liaoning Province Urban and Rural Construction and Planning Design Institute May 2017 Contents KEY PRINCIPLES OF RESETTLEMENT AND DEFINITIONS OF MAIN TERMS .......................................................................................................................... 1 1 OVERVIEW OF THE PROJECT ........................................................................... 4 1.1 OBJECTIVES OF THE PROJECT ............................................................................. 4 1.2 COMPONENTS OF THE PROJECT ........................................................................... 4 1.3 ALTERNATIVE DESIGN EXPLORED TO AVOID OR MINIMIZE RESETTLEMENT ... 10 1.4 LINKED PROJECTS IDENTIFICATION .................................................................. 10 2 PROJECT IMPACTS ............................................................................................. 13 2.1 IMPACT OF PERMANENT LAND ACQUISITION .................................................. -

Area Comprehensive Score 1990 2000 2010 Heping District 0.307
Comprehensive score of aging level in 1990, 2000 and 2010 Comprehensive score Area 1990 2000 2010 Heping District 0.307 0.572 0.792 Shenhe District 0.319 0.554 0.774 Dadong District 0.275 0.558 0.803 Huanggu District 0.262 0.542 0.777 Tiexi District (Shenyang) 0.252 0.611 0.800 Sujiatun District 0.202 0.409 0.699 Dongling District 0.202 0.370 0.512 Shenbei New District 0.196 0.388 0.534 Yuhong District 0.197 0.364 0.593 Liaozhong County 0.187 0.351 0.627 Kangping County 0.165 0.318 0.604 Faku County 0.195 0.354 0.653 Xinmin City 0.177 0.351 0.627 Zhongshan District 0.336 0.592 0.888 Xigang District 0.327 0.605 0.860 Shahekou District 0.284 0.534 0.770 Ganjingzi District 0.242 0.381 0.557 Lushunkou District 0.302 0.427 0.668 Jinzhou District 0.267 0.360 0.531 Changhai County 0.215 0.314 0.638 Wafangdian City 0.218 0.431 0.799 Pulandian City 0.243 0.440 0.812 Zhuanghe City 0.224 0.460 0.778 Tiedong District 0.230 0.541 0.831 Tiexi District (Anshan) 0.234 0.514 0.896 Lishan District 0.198 0.540 0.950 Qianshan District 0.215 0.399 0.721 Tai'an County 0.187 0.355 0.613 Xiuyan Manchu Autonomous County 0.171 0.349 0.620 Haicheng City 0.191 0.321 0.573 Xinfu District 0.245 0.517 0.853 Dongzhou District 0.230 0.551 1.000 Wanghua District 0.206 0.464 0.814 Shuncheng District 0.195 0.479 0.819 Fushun County 0.256 0.401 0.701 Xinbin Manchu Autonomous County 0.110 0.298 0.615 Qingyuan Manchu Autonomous County 0.124 0.318 0.618 Pingshan District 0.208 0.475 0.778 Xihu District 0.217 0.497 0.829 Mingshan District 0.186 0.440 0.743 Nanfen District 0.196 -

Asian Development Bank
Due Diligence Report August 2017 PRC: Integrated Development of Key Townships in Central Liaoning Project-Fuxin Old and Damaged Heat Network Energy-saving Reconstruction Subproject Prepared by the Government of Fuxin City for the Asian Development Bank. This due diligence report is a document of the borrower. The views expressed herein do not necessarily represent those of ADB's Board of Directors, Management, or staff, and may be preliminary in nature. Your attention is directed to the “terms of use” section of this website. In preparing any country program or strategy, financing any project, or by making any designation of or reference to a particular territory or geographic area in this document, the Asian Development Bank does not intend to make any judgments as to the legal or other status of any territory or area. Due Diligence Report People's Republic of China Fuxin Old and Damaged Heat Network Energy-Saving Reconstruction Project under Integrated Development of Key Townships in Central Liaoning Project (2901-PRC) Prepared for the Asian Development Bank (ADB) Prepared by the Government of Fuxin City Liaoning Daao Environmental Impact Assessment Co., Ltd August 2017 1 TABLE OF CONTENTS I General ............................................................................................................................................ 1 A. Overview ............................................................................................................................. 1 B. Laws, Regulations, Guidelines, Standards and Specifications -

Resettlement Plan
RESETTLEMENT PLAN LIAONING ENVIRONMENTAL IMPROVEMENT PROJECT IN THE PEOPLE’S REPUBLIC OF CHINA PROJECT MANAGEMENT OFFICE OF LIAONING ENVIRONMENT IMPROVEMENT AND FUXIN CBM/CMM DEVELOPMENT AND UTILIZATION COMPANY September 2004 THIS IS NOT AN ADB BOARD APPROVED DOCUMENT LIAONING ENVIRONMENTAL IMPROVEMENT PROJECT RESETTLEMENT PLAN 1. INTRODUCTION......................................................................................................................... 1 1.1 OBJECTIVE AND SCOPE OF THE PROJECT ................................................................................. 1 1.2 DESCRIPTIONS OF THE PROJECT .............................................................................................. 3 1.2.1 CBM/CMM Development Component ............................................................................ 3 1.2.2. Gas Distribution Component .......................................................................................... 3 1.2.3. City Central Heating Supply Component........................................................................ 4 1.3 TOTAL PROJECT INVESTMENT.................................................................................................. 5 2. IMPACTS OF LAND ACQUISITION AND RESETLEMENT ............................................... 6 2.1 ....................................................................................................................................................... 6 FUXIN CBM/CMM DEVELOPMENT PROJECT..................................................................................... -

A12 List of China's City Gas Franchising Zones
附录 A12: 中国城市管道燃气特许经营区收录名单 Appendix A03: List of China's City Gas Franchising Zones • 1 Appendix A12: List of China's City Gas Franchising Zones 附录 A12:中国城市管道燃气特许经营区收录名单 No. of Projects / 项目数:3,404 Statistics Update Date / 统计截止时间:2017.9 Source / 来源:http://www.chinagasmap.com Natural gas project investment in China was relatively simple and easy just 10 CNG)、控股投资者(上级管理机构)和一线运营单位的当前主官经理、公司企业 years ago because of the brand new downstream market. It differs a lot since 所有制类型和联系方式。 then: LNG plants enjoyed seller market before, while a LNG plant investor today will find himself soon fighting with over 300 LNG plants for buyers; West East 这套名录的作用 Gas Pipeline 1 enjoyed virgin markets alongside its paving route in 2002, while today's Xin-Zhe-Yue Pipeline Network investor has to plan its route within territory 1. 在基础数据收集验证层面为您的专业信息团队节省 2,500 小时之工作量; of a couple of competing pipelines; In the past, city gas investors could choose to 2. 使城市燃气项目投资者了解当前特许区域最新分布、其他燃气公司的控股势力范 sign golden areas with best sales potential and easy access to PNG supply, while 围;结合中国 LNG 项目名录和中国 CNG 项目名录时,投资者更易于选择新项 today's investors have to turn their sights to areas where sales potential is limited 目区域或谋划收购对象; ...Obviously, today's investors have to consider more to ensure right decision 3. 使 LNG 和 LNG 生产商掌握采购商的最新布局,提前为充分市场竞争做准备; making in a much complicated gas market. China Natural Gas Map's associated 4. 便于 L/CNG 加气站投资者了解市场进入壁垒,并在此基础上谨慎规划选址; project directories provide readers a fundamental analysis tool to make their 5. 结合中国天然气管道名录时,长输管线项目的投资者可根据竞争性供气管道当前 decisions. With a completed idea about venders, buyers and competitive projects, 格局和下游用户的分布,对管道路线和分输口建立初步规划框架。 analyst would be able to shape a better market model when planning a new investment or marketing program. -

Annual Development Report on China's Trademark Strategy 2013
Annual Development Report on China's Trademark Strategy 2013 TRADEMARK OFFICE/TRADEMARK REVIEW AND ADJUDICATION BOARD OF STATE ADMINISTRATION FOR INDUSTRY AND COMMERCE PEOPLE’S REPUBLIC OF CHINA China Industry & Commerce Press Preface Preface 2013 was a crucial year for comprehensively implementing the conclusions of the 18th CPC National Congress and the second & third plenary session of the 18th CPC Central Committee. Facing the new situation and task of thoroughly reforming and duty transformation, as well as the opportunities and challenges brought by the revised Trademark Law, Trademark staff in AICs at all levels followed the arrangement of SAIC and got new achievements by carrying out trademark strategy and taking innovation on trademark practice, theory and mechanism. ——Trademark examination and review achieved great progress. In 2013, trademark applications increased to 1.8815 million, with a year-on-year growth of 14.15%, reaching a new record in the history and keeping the highest a mount of the world for consecutive 12 years. Under the pressure of trademark examination, Trademark Office and TRAB of SAIC faced the difficuties positively, and made great efforts on soloving problems. Trademark Office and TRAB of SAIC optimized the examination procedure, properly allocated examiners, implemented the mechanism of performance incentive, and carried out the “double-points” management. As a result, the Office examined 1.4246 million trademark applications, 16.09% more than last year. The examination period was maintained within 10 months, and opposition period was shortened to 12 months, which laid a firm foundation for performing the statutory time limit. —— Implementing trademark strategy with a shift to effective use and protection of trademark by law.