Prevalence and Outcomes of Anemia and Hematinic Deficiencies in Patients with Chronic Heart Failure
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clinical Excellence in Cardiology
Clinical Excellence in Cardiology Roy C. Ziegelstein, MD* A recent study identified 7 domains of clinical excellence on the basis of interviews with “clinically excellent” physicians at academic institutions in the United States: (1) commu- nication and interpersonal skills, (2) professionalism and humanism, (3) diagnostic acu- men, (4) skillful negotiation of the health care system, (5) knowledge, (6) taking a scholarly approach to clinical practice, and (7) having passion for clinical medicine. What constitutes clinical excellence in cardiology has not previously been defined. The author discusses clinical excellence in cardiology using the framework of these 7 domains and also considers the additional domain of clinical experience. Specific aspects of the domains of clinical excellence that are of greatest relevance to cardiology are highlighted. In conclusion, this discussion characterizes what constitutes clinical excellence in cardiology and should stimulate additional discussion of the topic and an examination of how the domains of clinical excellence in cardiology are related to specific patient outcomes. © 2011 Elsevier Inc. All rights reserved. (Am J Cardiol 2011;108:607–611) On the basis of interviews with 24 academic physicians and clinical excellence has not been clearly demonstrated. deemed “clinically excellent,” Christmas et al1 identified 7 For example, the compliance of hospitals with performance domains of clinical excellence relevant to all disciplines in measures is not associated with improved heart failure out- medicine: (1) communication and interpersonal skills, (2) comes.10,11 The speed with which an interventional cardi- professionalism and humanism, (3) diagnostic acumen, (4) ologist achieves reperfusion of the culprit vessel, the so- skillful negotiation of the health care system, (5) knowl- called door-to-balloon time, is an important performance edge, (6) taking a scholarly approach to clinical practice, measure in the treatment of patients with acute ST-segment and (7) having passion for clinical medicine. -

Relevance of Hematological Parameters in Patients with Recurrent Aphthous Stomatitis
[Downloaded free from http://www.dmrjournal.org on Tuesday, October 22, 2019, IP: 83.34.92.213] Review Article Relevance of Hematological Parameters in Patients with Recurrent Aphthous Stomatitis Alberto Rodriguez‑Archilla, Maria Brykova Department of Stomatology, Oral Medicine Unit, Faculty of Dentistry, University of Granada, Granada, Spain Abstract Background: Recurrent aphthous stomatitis (RAS) continues to be a very common ulcerative disease of the oral mucosa, affecting approximately 20% of the general population. Hematinic deficiencies have been considered as a possible triggering factor, being iron, Vitamin 12B , or folic acid deficiencies two times more frequent in patients with RAS.Objective: The objective was to assess the hematological parameters as possible etiological factors of RAS. Materials and Methods: A PubMed search of articles on hematological parameters in RAS was conducted. From 93 articles published between 1954 and 2018 (64 with full-text availability), 45 were excluded for several reasons: studies without a control group (17), studies with no clinical data (12), and studies with nonusable data (16). Data were processed using the statistical software RevMan 5.3 (The Cochrane Collaboration, Oxford, UK). For continuous outcomes, the estimates of effects of an intervention were expressed as mean differences using the inverse variance method, and for dichotomous outcomes, the estimates of effects of an intervention were expressed as odds ratio (OR) using Mantel-Haenszel method, both with 95% confidence intervals.Results: Nineteen studies of hematological parameters on RAS were included in this meta-analysis. RAS patients had a significantly higher risk of presenting low levels, together with lower concentrations, of hemoglobin (OR: 17.30), iron (OR: 6.67), folic acid (OR: 4.98), Vitamin B12 (OR: 3.99), ferritin (OR: 2.86), and higher levels of homocysteine (OR: 7.22). -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -

A Distant Gene Deletion Affects Beta-Globin Gene Function in an Atypical Gamma Delta Beta-Thalassemia
A distant gene deletion affects beta-globin gene function in an atypical gamma delta beta-thalassemia. P Curtin, … , A D Stephens, H Lehmann J Clin Invest. 1985;76(4):1554-1558. https://doi.org/10.1172/JCI112136. Research Article We describe an English family with an atypical gamma delta beta-thalassemia syndrome. Heterozygosity results in a beta-thalassemia phenotype with normal hemoglobin A2. However, unlike previously described cases, no history of neonatal hemolytic anemia requiring blood transfusion was obtained. Gene mapping showed a deletion that extended from the third exon of the G gamma-globin gene upstream for approximately 100 kilobases (kb). The A gamma-globin, psi beta-, delta-, and beta-globin genes in cis remained intact. The malfunction of the beta-globin gene on a chromosome in which the deletion is located 25 kb away suggests that chromatin structure and conformation are important for globin gene expression. Find the latest version: https://jci.me/112136/pdf A Distant Gene Deletion Affects ,8-Globin Gene Function in an Atypical '6y5-Thalassemia Peter Curtin, Mario Pirastu, and Yuet Wai Kan Howard Hughes Medical Institute and Department ofMedicine, University of California, San Francisco, California 94143 John Anderson Gobert-Jones Department ofPathology, West Suffolk County Hospital, Bury St. Edmunds IP33-2QZ, Suffolk, England Adrian David Stephens Department ofHaematology, St. Bartholomew's Hospital, London ECIA-7BE, England Herman Lehmann Department ofBiochemistry, University ofCambridge, Cambridge CB2-lQW, England Abstract tologic picture of f3-thalassemia minor in adult life. Globin syn- thetic studies reveal a ,3 to a ratio of -0.5, but unlike the usual We describe an English family with an atypical 'yS6-thalassemia fl-thalassemia heterozygote, the levels of HbA2 (and HbF) are syndrome. -

Anemia in Heart Failure - from Guidelines to Controversies and Challenges
52 Education Anemia in heart failure - from guidelines to controversies and challenges Oana Sîrbu1,*, Mariana Floria1,*, Petru Dascalita*, Alexandra Stoica1,*, Paula Adascalitei, Victorita Sorodoc1,*, Laurentiu Sorodoc1,* *Grigore T. Popa University of Medicine and Pharmacy; Iasi-Romania 1Sf. Spiridon Emergency Hospital; Iasi-Romania ABSTRACT Anemia associated with heart failure is a frequent condition, which may lead to heart function deterioration by the activation of neuro-hormonal mechanisms. Therefore, a vicious circle is present in the relationship of heart failure and anemia. The consequence is reflected upon the pa- tients’ survival, quality of life, and hospital readmissions. Anemia and iron deficiency should be correctly diagnosed and treated in patients with heart failure. The etiology is multifactorial but certainly not fully understood. There is data suggesting that the following factors can cause ane- mia alone or in combination: iron deficiency, inflammation, erythropoietin levels, prescribed medication, hemodilution, and medullar dysfunc- tion. There is data suggesting the association among iron deficiency, inflammation, erythropoietin levels, prescribed medication, hemodilution, and medullar dysfunction. The main pathophysiologic mechanisms, with the strongest evidence-based medicine data, are iron deficiency and inflammation. In clinical practice, the etiology of anemia needs thorough evaluation for determining the best possible therapeutic course. In this context, we must correctly treat the patients’ diseases; according with the current guidelines we have now only one intravenous iron drug. This paper is focused on data about anemia in heart failure, from prevalence to optimal treatment, controversies, and challenges. (Anatol J Cardiol 2018; 20: 52-9) Keywords: anemia, heart failure, intravenous iron, ferric carboxymaltose, quality of life Introduction g/dL in men) (2). -

Eosinophilic Gastroenteritis Presenting with Severe Anemia and Near Syncope
J Am Board Fam Med: first published as 10.3122/jabfm.2012.06.110269 on 7 November 2012. Downloaded from BRIEF REPORT Eosinophilic Gastroenteritis Presenting with Severe Anemia and Near Syncope Nneka Ekunno, DO, MPH, Kirk Munsayac, DO, Allen Pelletier, MD, and Thad Wilkins, MD Eosinophilic gastrointestinal disorders or eosinophilic digestive disorders encompass a spectrum of rare gastrointestinal disorders that includes eosinophilic esophagitis, eosinophilic gastroenteritis, and eosinophilic colitis. Eosinophilic gastroenteritis is a rare inflammatory disease characterized by eosino- philic infiltration of the gastrointestinal tract. The clinical manifestations include anemia, dyspepsia, and diarrhea. Endoscopy with biopsy showing histologic evidence of eosinophilic infiltration is consid- ered definitive for diagnosis. Corticosteroid therapy, food allergen testing, elimination diets, and ele- mental diets are considered effective treatments for eosinophilic gastroenteritis. The treatment and prognosis of eosinophilic gastroenteritis is determined by the severity of the clinical manifestations. We describe a 24-year-old woman with eosinophilic gastroenteritis presenting as epigastric pain with a history of severe iron deficiency anemia, asthma, eczema, and allergic rhinitis, and we review the litera- ture regarding presentation, diagnostic testing, pathophysiology, predisposing factors, and treatment recommendations. (J Am Board Fam Med 2012;25:913–918.) Keywords: Case Reports, Eosinophilic Gastroenteritis, Gastrointestinal Disorders copyright. A 24-year-old nulliparous African-American woman During examination, her height was 62 inches, was admitted after an episode of near syncope asso- weight 117 lb, and body mass index 21.44 kg/m2. Her ciated with 2 days of fatigue and dizziness. She re- heart rate was 111 beats per minute, blood pressure ported gradual onset of dyspepsia over 2 to 3 121/57 mm Hg, respiratory rate 20 breaths per minute, months. -

Training in Nuclear Cardiology
JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY VOL.65,NO.17,2015 ª 2015 BY THE AMERICAN COLLEGE OF CARDIOLOGY FOUNDATION ISSN 0735-1097/$36.00 PUBLISHED BY ELSEVIER INC. http://dx.doi.org/10.1016/j.jacc.2015.03.019 TRAINING STATEMENT COCATS 4 Task Force 6: Training in Nuclear Cardiology Endorsed by the American Society of Nuclear Cardiology Vasken Dilsizian, MD, FACC, Chair Todd D. Miller, MD, FACC James A. Arrighi, MD, FACC* Allen J. Solomon, MD, FACC Rose S. Cohen, MD, FACC James E. Udelson, MD, FACC, FASNC 1. INTRODUCTION ACC and ASNC, and addressed their comments. The document was revised and posted for public comment 1.1. Document Development Process from December 20, 2014, to January 6, 2015. Authors 1.1.1. Writing Committee Organization addressed additional comments from the public to complete the document. The final document was The Writing Committee was selected to represent the approved by the Task Force, COCATS Steering Com- American College of Cardiology (ACC) and the Amer- mittee, and ACC Competency Management Commit- ican Society of Nuclear Cardiology (ASNC) and tee; ratified by the ACC Board of Trustees in March, included a cardiovascular training program director; a 2015; and endorsed by the ASNC. This document is nuclear cardiology training program director; early- considered current until the ACC Competency Man- career experts; highly experienced specialists in agement Committee revises or withdraws it. both academic and community-based practice set- tings; and physicians experienced in defining and applying training standards according to the 6 general 1.2. Background and Scope competency domains promulgated by the Accredita- Nuclear cardiology provides important diagnostic and tion Council for Graduate Medical Education (ACGME) prognostic information that is an essential part of the and American Board of Medical Specialties (ABMS), knowledge base required of the well-trained cardiol- and endorsed by the American Board of Internal ogist for optimal management of the cardiovascular Medicine (ABIM). -

Essentials of Cardiology Timothy C
Th e Heart SECTION IV Essentials of Cardiology Timothy C. Slesnick, Ralph Gertler, and CHAPTER 14 Wanda C. Miller-Hance Congenital Heart Disease Evaluation of the Patient with a Cardiac Murmur Incidence Basic Interpretation of the Pediatric Segmental Approach to Diagnosis Electrocardiogram Physiologic Classifi cation of Defects Essentials of Cardiac Rhythm Interpretation and Acute Arrhythmia Management in Children Acquired Heart Disease Basic Rhythms Cardiomyopathies Conduction Disorders Myocarditis Cardiac Arrhythmias Rheumatic Fever and Rheumatic Heart Disease Pacemaker Therapy in the Pediatric Age Group Infective Endocarditis Pacemaker Nomenclature Kawasaki Disease Permanent Cardiac Pacing Cardiac Tumors Diagnostic Modalities in Pediatric Cardiology Heart Failure in Children Chest Radiography Defi nition and Pathophysiology Barium Swallow Etiology and Clinical Features Echocardiography Treatment Strategies Magnetic Resonance Imaging Syndromes, Associations, and Systemic Disorders: Cardiovascular Disease and Anesthetic Implications Computed Tomography Chromosomal Syndromes Cardiac Catheterization and Angiography Gene Deletion Syndromes Considerations in the Perioperative Care of Children with Cardiovascular Disease Single-Gene Defects General Issues Associations Clinical Condition and Status of Prior Repair Other Disorders Summary Selected Vascular Anomalies and Their Implications for Anesthetic Care Aberrant Subclavian Arteries Persistent Left Superior Vena Cava to Coronary Sinus Communication Congenital Heart Disease adulthood has become the expectation for most congenital car- diovascular malformations.3 At present it is estimated that there Incidence are more than a million adults with CHD in the United States, Estimates of the incidence of congenital heart disease (CHD) surpassing the number of children similarly aff ected for the fi rst range from 0.3% to 1.2% in live neonates.1 Th is represents the time in history. -

Bed-Based Ballistocardiography: Dataset and Ability to Track Cardiovascular Parameters
sensors Article Bed-Based Ballistocardiography: Dataset and Ability to Track Cardiovascular Parameters Charles Carlson 1,* , Vanessa-Rose Turpin 2, Ahmad Suliman 1 , Carl Ade 2, Steve Warren 1 and David E. Thompson 1 1 Mike Wiegers Department of Electrical and Computer Engineering, Kansas State University, Manhattan, KS 66506, USA; [email protected] (A.S.); [email protected] (S.W.); [email protected] (D.E.T.) 2 Department of Kinesiology, Kansas State University, Manhattan, KS 66506, USA; [email protected] (V.-R.T.); [email protected] (C.A.) * Correspondence: [email protected] Abstract: Background: The goal of this work was to create a sharable dataset of heart-driven signals, including ballistocardiograms (BCGs) and time-aligned electrocardiograms (ECGs), photoplethysmo- grams (PPGs), and blood pressure waveforms. Methods: A custom, bed-based ballistocardiographic system is described in detail. Affiliated cardiopulmonary signals are acquired using a GE Datex CardioCap 5 patient monitor (which collects ECG and PPG data) and a Finapres Medical Systems Finometer PRO (which provides continuous reconstructed brachial artery pressure waveforms and derived cardiovascular parameters). Results: Data were collected from 40 participants, 4 of whom had been or were currently diagnosed with a heart condition at the time they enrolled in the study. An investigation revealed that features extracted from a BCG could be used to track changes in systolic blood pressure (Pearson correlation coefficient of 0.54 +/− 0.15), dP/dtmax (Pearson correlation coefficient of 0.51 +/− 0.18), and stroke volume (Pearson correlation coefficient of 0.54 +/− 0.17). Conclusion: A collection of synchronized, heart-driven signals, including BCGs, ECGs, PPGs, and blood pressure waveforms, was acquired and made publicly available. -

Alpha Thalassemia Trait
Alpha Thalassemia Trait Alpha Thalassemia Trait Produced by St. Jude Children’s Research Hospital, Departments of Hematology, Patient Education, 1 and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to replace counseling by a trained health care professional or genetic counselor. Our aim is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specific treatment options should be discussed with your doctor. For general information on sickle cell disease and other blood disorders, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital Alpha thalassemia trait All red blood cells contain hemoglobin (HEE muh glow bin), which carries oxygen from your lungs to all parts of your body. Alpha thalassemia (thal uh SEE mee uh) trait is a condition that affects the amount of hemo- globin in the red blood cells. • Adult hemoglobin (hemoglobin A) is made of alpha and beta globins. • Normally, people have 4 genes for alpha globin with 2 genes on each chromosome (aa/aa). People with alpha thalassemia trait only have 2 genes for alpha globin, so their bodies make slightly less hemoglobin than normal. This trait was passed on from their parents, like hair color or eye color. A trait is different from a disease 2 Alpha thalassemia trait is not a disease. Normally, a trait will not make you sick. Parents who have alpha thalassemia trait can pass it on to their children. -
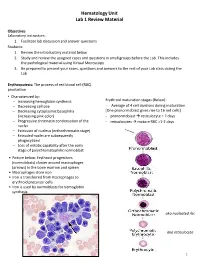
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Molecular Epidemiology and Hematologic Characterization of Δβ
Jiang et al. BMC Medical Genetics (2020) 21:43 https://doi.org/10.1186/s12881-020-0981-x RESEARCH ARTICLE Open Access Molecular epidemiology and hematologic characterization of δβ-thalassemia and hereditary persistence of fetal hemoglobin in 125,661 families of greater Guangzhou area, the metropolis of southern China Fan Jiang1,2, Liandong Zuo2, Dongzhi Li2, Jian Li2, Xuewei Tang2, Guilan Chen2, Jianying Zhou2, Hang Lu2 and Can Liao1,2* Abstract Background: Individuals with δβ-thalassemia/HPFH and β-thalassemia usually present with intermedia or thalassemia major. No large-scale survey on HPFH/δβ-thalassemia in southern China has been reported to date. The purpose of this study was to examine the molecular epidemiology and hematologic characteristics of these disorders in Guangzhou, the largest city in Southern China, to offer advice for thalassemia screening programs and genetic counseling. Methods: A total of 125,661 couples participated in pregestational thalassemia screening. 654 subjects with fetal hemoglobin (HbF) level ≥ 5% were selected for further investigation. Gap-PCR combined with Multiplex ligation dependent probe amplification (MLPA) was used to screen for β-globin gene cluster deletions. Gene sequencing for the promoter region of HBG1 /HBG2 gene was performed for all those subjects. Results: A total of 654 individuals had hemoglobin (HbF) levels≥5, and 0.12% of the couples were found to be heterozygous for HPFH/δβ-thalassemia, including Chinese Gγ (Aγδβ)0-thal, Southeast Asia HPFH (SEA-HPFH), Taiwanese deletion and Hb Lepore–Boston–Washington. The highest prevalence was observed in the Huadu district and the lowest in the Nansha district. Three cases were identified as carrying β-globin gene cluster deletions, which had not been previously reported.