NUCLEAR WASTE PROCESSING BASED on FOOF and Krf 2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Material Safety Data Sheet Prepared to US OSHA, CMA, ANSI, Canadian WHMIS and EU Standards
MSDS NUMBER: 1201 Fluorine Excimer Laser Mix (1.0% or less Fluorine) Material Safety Data Sheet Prepared to US OSHA, CMA, ANSI, Canadian WHMIS and EU Standards SECTION 1. PRODUCT IDENTIFICATION PRODUCT NAME: Fluorine Excimer Laser Mix (1.0% or less Fluorine) US DOT NAME: Compressed Gas, n.o.s. (Fluorine, other gas) UN 1956 (see Sec. 14) CHEMICAL NAME: Mixture of Fluorine (1.0% or Less) and Argon, Krypton or Xenon balance Helium, Neon or Nitrogen FORMULA: Argon = Ar; Fluorine = F2; Helium = He; Krypton = Kr; Neon = Ne; Nitrogen = N2; SYNONYMS: Not Applicable US MANUFACTURER: Linde LLC 575 Mountain Ave. Murray Hill, NJ 07974 Phone: 908-464-8100 lindeus.com 24 HOUR EMERGENCY CONTACT, CHEMTREC: 800/424-9300, 703/527-3887 For additional product information contact your customer service representative. PRODUCT USE: In Excimer Laser and Research and Development ALL WHMIS required information is included in appropriate sections based on the ANSI Z400.1-2004 format. This product has been classified in accordance with the hazard criteria of the CPR and the MSDS contains all the information required by the CPR. The product is also classified per all applicable EU Directives through EC 1907: 2006 SECTION 2. HAZARD IDENTIFICATION EU LABELING AND CLASSIFICATION: This gas mixture meets the definition of hazardous according to the criteria of the European Union Council Directive 67/548/EEC and based on current guidelines under EC 1907: 2006. EU CLASSIFICATION: Xn (Harmful) EU RISK PHRASES: R: 20; R: 21; R: 36/37/38 EU SAFETY PHRASES: S: (1/2)*; S: 7/9, S: 26, S: 36/37/39, S: 45 See Section 15 for full definition of Risk and Safety Phrases. -

1. Trioxygen Difluoride Reacts with Fluorine to Form A. Dioxygen Trifluoride B
1. Trioxygen difluoride reacts with fluorine to form a. Dioxygen trifluoride b. Dioxygen difluoride c. Oxygen difluoride d. Oxygen & fluorine 2. I2O4 and I2O9 are salt like and a. Are considered as iodine salt b. Are used in table salt c. Are called as iodine – iodates d. None of them 3. Reaction of HClO4 with P2O5 produces a. Chlorine heptaoxide b. Chlorine dioxide c. Dichlorine monoxide d. Chlorine hexaoxide 4. Reaction of bromine vapours with mercuric oxide yields a. Bromine dioxide b. Bromine trioxide c. Bromine monoxide d. All of them 5. Iodine pentaoxide acts as a. An oxidizing agent b. Reducing agent c. Bleaching agent d. Dehydrating agent 6. HClO2 is called a. Hypochlorous acid b. Chlorous acid c. Perchloric acid d. None of them 7. Which compound is used for the quantitative analysis of CO? a. O3F2 b. ClO2 c. Br2O d. I2O5 8. Bleaching powder can be manufactured by a. Hasenclever’s method b. Beckmann’s method c. Both a & b d. None of them 9. Bleaching powder oxidizes NH3 & yields a. Chlorine gas b. Nitrogen gas c. NH4Cl d. None of them 10. Freon is the commercial name of a. fluorochlorcarbons b. Tetrafluoroethylene c. Fluoroethylene d. None of them 11. Halothane is used as a. Disinfectant b. Anaesthetic c. Fungicicle d. Insecticide 12. Which one of the following halides has the highest lattlice energies? a. Iodides b. Chlorides c. Fluorides d. None of them 13. Which one of the following Halogens has the highest oxidizing power? a. F b. Cl c. I d. Br 14. Which one of the following Halogens can oxidize all other halide ions to molecular Halogen? a. -

Chemical Chemical Hazard and Compatibility Information
Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 degrees F) to avoid rupture of carboys and glass containers.. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino-ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers (strong), P(OCN)3, Perchloric acid, Permanganates, Peroxides, Phenols, Phosphorus isocyanate, Phosphorus trichloride, Potassium hydroxide, Potassium permanganate, Potassium-tert-butoxide, Sodium hydroxide, Sodium peroxide, Sulfuric acid, n-Xylene. Acetone HAZARDS & STORAGE: Store in a cool, dry, well ventilated place. INCOMPATIBILITIES: Acids, Bromine trifluoride, Bromine, Bromoform, Carbon, Chloroform, Chromium oxide, Chromium trioxide, Chromyl chloride, Dioxygen difluoride, Fluorine oxide, Hydrogen peroxide, 2-Methyl-1,2-butadiene, NaOBr, Nitric acid, Nitrosyl chloride, Nitrosyl perchlorate, Nitryl perchlorate, NOCl, Oxidizing materials, Permonosulfuric acid, Peroxomonosulfuric acid, Potassium-tert-butoxide, Sulfur dichloride, Sulfuric acid, thio-Diglycol, Thiotrithiazyl perchlorate, Trichloromelamine, 2,4,6-Trichloro-1,3,5-triazine -

H2O2 and NH 2 OH
Int. J. Mol. Sci. 2006 , 7, 289-319 International Journal of Molecular Sciences ISSN 1422-0067 © 2006 by MDPI www.mdpi.org/ijms/ A Quest for the Origin of Barrier to the Internal Rotation of Hydrogen Peroxide (H 2O2) and Fluorine Peroxide (F 2O2) Dulal C. Ghosh Department of Chemistry, University of Kalyani, Kalyani-741235, India Email: [email protected], Fax: +91-33 25828282 Received: 2 July 2006 / Accepted: 24 July 2006 / Published: 25 August 2006 Abstract: In order to understand the structure-property relationship, SPR, an energy- partitioning quest for the origin of the barrier to the internal rotation of two iso-structural molecules, hydrogen peroxide, H 2O2, and fluorine peroxide, F 2O2 is performed. The hydrogen peroxide is an important bio-oxidative compound generated in the body cells to fight infections and is an essential ingredient of our immune system. The fluorine peroxide is its analogue. We have tried to discern the interactions and energetic effects that entail the nonplanar skew conformation as the equilibrium shape of the molecules. The physical process of the dynamics of internal rotation initiates the isomerization reaction and generates infinite number of conformations. The decomposed energy components faithfully display the physical process of skewing and eclipsing as a function of torsional angles and hence are good descriptors of the process of isomerization reaction of hydrogen peroxide (H 2O2) and dioxygen difluoride (F 2O2) associated with the dynamics of internal rotation. It is observed that the one-center, two-center bonded and nonbonded interaction terms are sharply divided in two groups. One group of interactions hinders the skewing and favours planar cis/trans forms while the other group favours skewing and prefers the gauche conformation of the molecule. -

19770005666.Pdf
General Disclaimer One or more of the Following Statements may affect this Document This document has been reproduced from the best copy furnished by the organizational source. It is being released in the interest of making available as much information as possible. This document may contain data, which exceeds the sheet parameters. It was furnished in this condition by the organizational source and is the best copy available. This document may contain tone-on-tone or color graphs, charts and/or pictures, which have been reproduced in black and white. This document is paginated as submitted by the original source. Portions of this document are not fully legible due to the historical nature of some of the material. However, it is the best reproduction available from the original submission. Produced by the NASA Center for Aerospace Information (CASI) V NASA TECHNICAL NASA TM X-73983 MEMORANDUM 00 X I-- A SURVEY OF KINETIC DATA OF COMPOUNDS CONTAINING FLUORINE Dana A. Brewer Langley Research Center (NASA-TM-X-73983) A SURVEY OF KINETIC DATA N77-12609 OF COMPOUNDS CONTAINING FLOURINE (NASA) 119 p, HC A06/MF A01 CSCL 04A Unclas G3/46 55838 November 1976 This informal documentation medium is used to provide accelerated or special release of technical information to selected users. The contents may not meet NASA formal editing and publication standards, may be re- vised, or may I be incorporated in another publication. 4WAil NATIONAL AERONAUTICS AND SPACE ADMINISTRATION DEC 1976 LANGLEY RESEARCH CENTER, HAMPTON, VIRGINIA 23665 WEIVED 0M tosp, Sn PACILITY INpUT BRANCH < C' ' r ^| ' 1. Report No. -

Material Safety Data Sheet Ethyl Alcohol 200 Proof MSDS
Material Safety Data Sheet Ethyl alcohol 200 Proof MSDS Section 1: Chemical Product and Company Identification Product Name: Ethyl alcohol 200 Proof Contact Information: Catalog Codes: SLE2248, SLE1357 Sciencelab.com, Inc. 14025 Smith Rd. CAS#: 64-17-5 Houston, Texas 77396 US Sales: 1-800-901-7247 RTECS: KQ6300000 International Sales: 1-281-441-4400 TSCA: TSCA 8(b) inventory: Ethyl alcohol 200 Proof Order Online: ScienceLab.com CI#: Not applicable. CHEMTREC (24HR Emergency Telephone), call: 1-800-424-9300 Synonym: Ethanol; Absolute Ethanol; Alcohol; Ethanol 200 proof; Ethyl Alcohol, Anhydrous; Ethanol, undenatured; International CHEMTREC, call: 1-703-527-3887 Dehydrated Alcohol; Alcohol For non-emergency assistance, call: 1-281-441-4400 Chemical Name: Ethyl Alcohol Chemical Formula: CH3CH2OH Section 2: Composition and Information on Ingredients Composition: Name CAS # % by Weight Ethyl alcohol 200 Proof 64-17-5 100 Toxicological Data on Ingredients: Ethyl alcohol 200 Proof: ORAL (LD50): Acute: 7060 mg/kg [Rat]. 3450 mg/kg [Mouse]. VAPOR (LC50): Acute: 20000 ppm 8 hours [Rat]. 39000 mg/m 4 hours [Mouse]. Section 3: Hazards Identification Potential Acute Health Effects: Hazardous in case of skin contact (irritant), of eye contact (irritant), of inhalation. Slightly hazardous in case of skin contact (permeator), of ingestion. Potential Chronic Health Effects: Slightly hazardous in case of skin contact (sensitizer). CARCINOGENIC EFFECTS: A4 (Not classifiable for human or animal.) by ACGIH. MUTAGENIC EFFECTS: Mutagenic for mammalian somatic cells. Mutagenic for bacteria and/or yeast. TERATOGENIC EFFECTS: Classified PROVEN for human. DEVELOPMENTAL TOXICITY: Classified Development toxin [PROVEN]. Classified Reproductive system/toxin/female, Reproductive system/toxin/male [POSSIBLE]. -

List of Reactive Chemicals
LIST OF REACTIVE CHEMICALS Chemical Prefix Chemical Name Reactive Reactive Reactive CAS# Chemical Chemical Chemical Stimulus 1 Stimulus 2 Stimulus 3 111-90-0 "CARBITOL" SOLVENT D 111-15-9 "CELLOSOLVE" ACETATE D 110-80-5 "CELLOSOLVE" SOLVENT D 2- (2,4,6-TRINITROPHENYL)ETHYL ACETATE (1% IN ACETONE & BENZENE S 12427-38-2 AAMANGAN W 88-85-7 AATOX S 40487-42-1 AC 92553 S 105-57-7 ACETAL D 75-07-0 ACETALDEHYDE D 105-57-7 ACETALDEHYDE, DIETHYL ACETAL D 108-05-4 ACETIC ACID ETHENYL ESTER D 108-05-4 ACETIC ACID VINYL ESTER D 75-07-0 ACETIC ALDEHYDE D 101-25-7 ACETO DNPT T 126-84-1 ACETONE DIETHYL ACETAL D 108-05-4 ACETOXYETHYLENE D 108-05-4 1- ACETOXYETHYLENE D 37187-22-7 ACETYL ACETONE PEROXIDE, <=32% AS A PASTE T 37187-22-7 ACETYL ACETONE PEROXIDE, <=42% T 37187-22-7 ACETYL ACETONE PEROXIDE, >42% T S 644-31-5 ACETYL BENZOYL PEROXIDE (SOLID OR MORE THAN 45% IN SOLUTION) T S 644-31-5 ACETYL BENZOYL PEROXIDE, <=45% T 506-96-7 ACETYL BROMIDE W 75-36-5 ACETYL CHLORIDE W ACETYL CYCLOHEXANE SULFONYL PEROXIDE (>82% WITH <12% WATER) T S 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=32% T 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=82% T 674-82-8 ACETYL KETENE (POISON INHALATION HAZARD) D 110-22-5 ACETYL PEROXIDE, <=27% T 110-22-5 ACETYL PEROXIDE, SOLID, OR MORE THAN 27% IN SOLUTION T S 927-86-6 ACETYLCHOLINE PERCHLORATE O S 74-86-2 ACETYLENE D 74-86-2 ACETYLENE (LIQUID) D ACETYLENE SILVER NITRATE D 107-02-08 ACRALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACROLEIC ACID D 107-02-08 ACROLEIN, INHIBITED (POISON INHALATION HAZARD) D 107-02-08 ACRYLALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACRYLIC ACID D 141-32-2 ACRYLIC ACID BUTYL ESTER D 140-88-5 ACRYLIC ACID ETHYL ESTER D 96-33-3 ACRYLIC ACID METHYL ESTER D Stimulus - Stimuli is the thermal, physical or chemical input needed to induce a hazardous reaction. -

Inorganic Chemistry
INORGANIC CHEMISTRY Saito Inorganic Chemistry Saito This text is disseminated via the Open Education Resource (OER) LibreTexts Project (https://LibreTexts.org) and like the hundreds of other texts available within this powerful platform, it freely available for reading, printing and "consuming." Most, but not all, pages in the library have licenses that may allow individuals to make changes, save, and print this book. Carefully consult the applicable license(s) before pursuing such effects. Instructors can adopt existing LibreTexts texts or Remix them to quickly build course-specific resources to meet the needs of their students. Unlike traditional textbooks, LibreTexts’ web based origins allow powerful integration of advanced features and new technologies to support learning. The LibreTexts mission is to unite students, faculty and scholars in a cooperative effort to develop an easy-to-use online platform for the construction, customization, and dissemination of OER content to reduce the burdens of unreasonable textbook costs to our students and society. The LibreTexts project is a multi-institutional collaborative venture to develop the next generation of open-access texts to improve postsecondary education at all levels of higher learning by developing an Open Access Resource environment. The project currently consists of 13 independently operating and interconnected libraries that are constantly being optimized by students, faculty, and outside experts to supplant conventional paper-based books. These free textbook alternatives are organized within a central environment that is both vertically (from advance to basic level) and horizontally (across different fields) integrated. The LibreTexts libraries are Powered by MindTouch® and are supported by the Department of Education Open Textbook Pilot Project, the UC Davis Office of the Provost, the UC Davis Library, the California State University Affordable Learning Solutions Program, and Merlot. -
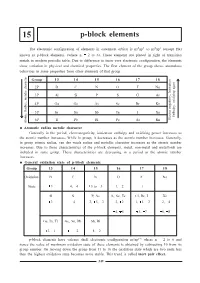
P-Block Elements
15 p-block elements The electronic configuration of elements in outermost orbital is ns2np1 to ns2np6 (except He) known as p-block elements. (where n = 2 to 6). These elements are placed in right of transition metals in modern periodic table. Due to difference in inner core electronic configuration, the elements show variation in physical and chemical properties. The first element of the group shows anomalous behaviour in some properties from other elements of that group. Group 13 14 15 16 17 18 2P BC NO F Ne 3P Al Si P S Cl Ar , Ionisation 4P Ga Ge As Se Br Kr , oxidizing agent 5P In Sn Sb Te I Xe Enthalpy 6P Tl Pb Bi Po At Rn Electro negativity Atomic Radius, metalic character Atomatic radius metalic character Generally in the period, electronegativity, ionization enthalpy and oxidising power increases as the atomic number increases. While in group, it decreases as the atomic number increases. Generally, in group atomic radius, van der waals radius and metallic character increases as the atomic number increases. Due to these characteristics of the p-block elements, metal, non-metal and metalloids are included in same group. These characteristics are decreasing in a period as the atomic number increases. General oxidation state of p-block elements Group 13 14 15 16 17 18 Oxidation BC NO F Ne State +3 +4, -4 +5 to -3 -1, -2 -1 - Al Si P, As S, Se, Te Cl, Br, I Xe +3 +4 +3, +5, -3 -2, +2 -1, +1, +3 +2, +4 +4, +6 +5, +7 +6, +8 Ga, In, Tl Ge, Sn, Pb Sb, Bi +3, +1 +4, +2 +4, +2 p-block elements have valence shell electronic configuration ns2np16 where n = 2 to 6 and hence the value of maximum oxidation state of these elements is obtained by subtracting 10 from its group number. -

Witch Hazel Extract, Double Distilled
SAFETY DATA SHEET Preparation Date: 11/11/2015 Revision Date: 11/11/2015 Revision Number: G1 1. IDENTIFICATION Product identifier Product code: WI105 Product Name: WITCH HAZEL EXTRACT, DOUBLE DISTILLED Other means of identification Synonyms: Amamelide, extract Hamamelis virginiana, extract CAS #: Mixture RTECS # MG8328500 CI#: Not available Recommended use of the chemical and restrictions on use Recommended use: No information available. Uses advised against No information available Supplier: Spectrum Chemical Mfg. Corp 14422 South San Pedro St. Gardena, CA 90248 (310) 516-8000 Order Online At: https://www.spectrumchemical.com Emergency telephone number Chemtrec 1-800-424-9300 Contact Person: Martin LaBenz (West Coast) Contact Person: Ibad Tirmiz (East Coast) 2. HAZARDS IDENTIFICATION Classification This chemical is considered hazardous by the 2012 OSHA Hazard Communication Standard (29 CFR 1910.1200) Serious eye damage/eye irritation Category 2B Flammable liquids Category 3 Label elements Product code: WI105 Product name: WITCH HAZEL 1 / 14 EXTRACT, DOUBLE DISTILLED Warning Hazard statements Causes eye irritation Flammable liquid and vapor Hazards not otherwise classified (HNOC) Not Applicable Other hazards Can burn with an invisible flame Precautionary Statements - Prevention Wash face, hands and any exposed skin thoroughly after handling Keep away from heat/sparks/open flames/hot surfaces. — No smoking Keep container tightly closed Ground/bond container and receiving equipment Use explosion-proof electrical/ventilating/lighting/ .? /equipment Use only non-sparking tools Take precautionary measures against static discharge Wear protective gloves Wear eye/face protection In case of fire: Use CO2, dry chemical, or foam to extinguish. IF IN EYES: Rinse cautiously with water for several minutes. -

=« Y * MASS SPECTROMETRIC STUDIES of the SYNTHESIS
"In presenting the dissertation as a partial fulfillment of the requirements for an advanced degree from the Georgia Institute of Technology, I agree that the Library of the Institution shall make it available for inspection and circulation in accordance with its regulations governing materials of this type. I agree that permission to copy from, or to publish from, this dissertation may be granted by the professor under whose direction it was written, or, in his absence, by the dean of the Graduate Division when such copying or publication is solely for scholarly purposes and does not involve potential financial gain. It is understood that any copying from, or publication of, this dissertation which involves potential financial gain will not be allowed without written permission, "-- -. • i -=« y * MASS SPECTROMETRIC STUDIES OF THE SYNTHESIS, REACTIVITY, AND ENERGETICS OF THE OXYGEN FLUORIDES AT CRYOGENIC TEMPERATURES A THESIS Presented to The Faculty of the Graduate Division by Thomas Joseph Malone In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the School of Chemical Engineering Georgia Institute of Technology February, 1966 MASS SPECTROMETRIC STUDIES OF THE SYNTHESIS, REACTIVITY, AND ENERGETICS OF THE OXYGEN FLUORIDES AT CRYOGENIC TEMPERATURES Approved; / • ' "/v Date approved by Chairma n:\PuJl 3 lUt, 11 ACKNOWLEDGMENTS I am deeply indebted to Dr0 Ho Ao McGee, Jr0 for his encouragement, advice^ and assistance throughout both my graduate and undergraduate studieso Many hours of discussion and willing -

P-BLOCK ELEMENTS and THEIR COMPOUNDS - II
MODULE - 6 Chemistry Chemistry of Elements 22 Notes p-BLOCK ELEMENTS AND THEIR COMPOUNDS - II You have already studied the chemistry of the elements of Groups 13, 14 and 15. In this lesson we shall deal with the chemistry of the elements of Groups 16, 17 and 18. Objectives After reading this lesson you will be able to: classify oxides into acidic, basic and amphoteric types; describe the manufacture of sulphuric acid; recall the preparation, properties and uses of ozone; recall the characteristics of hydrogen halides (HF, HCl); list the oxides and oxoacids of chlorine; compare the acidic behaviour of oxoacids of chlorine; write the general molecular formulae of interhalogen compounds; discuss the structures of interhalogen compounds; list a few chloro fluoro carbons and explain their uses and their effect on environment; explain the unreactive nature of noble gases; recall the preparation of xenon fluorides and oxides, and illustrate the structures of XeF2, XeF4, XeF6, XeO3 and XeO4. 22.1 Oxygen and Sulphur Oxygen and sulphur are the first two members of the 16th group of the periodic table. In this section you will learn about some compounds of oxygen and sulphur including environmentally important ozone and industrially important sulphuric acid. 62 p-Block Elements and Their Compounds - II MODULE - 6 22.1.1. Classification of Oxides Chemistry of Elements The binary compounds of oxygen with other elements (metals or non-metals) are called oxides. An understanding of the nature of an oxide provides a clue to the nature of the element which forms the oxide. Depending upon the acid-base behaviour of the oxides, they can be classified into the following categories.