Short-Limbed Dwarfism with Bowing, Combined Immune Deficiency, and Late Onset Aplastic Anaemia Caused by Novel Mutations In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metaphyseal Dysplasia: a Rare Case Report Dildip Khanal* Karuna Foundation Nepal “Saving Children from Disability, One by One”, Nepal
ical C lin as Khanal, J Clin Case Rep 2016, 6:2 C e f R o l e a p DOI: 10.4172/2165-7920.1000726 n o r r t u s o J Journal of Clinical Case Reports ISSN: 2165-7920 Case Report Open Access Metaphyseal Dysplasia: A Rare Case Report Dildip Khanal* Karuna Foundation Nepal “Saving Children from Disability, One by One”, Nepal Abstract Metaphyseal dysplasia is a very rare inherited bone disorder. Here is a case report and possible treatment options for 11 years old child, detected by Karuna foundation Nepal. Keywords: Metaphyseal dysplasia; Pyle; Therapeutic rehabilitation; Karuna foundation Nepal Background Metaphyseal dysplasia also known as Pyle disease is a heterogeneous group of disorders, characterized by the metaphyseal changes of the tubular bones with normal epiphyses. The disease was described briefly by Pyle in 1931 [1,2]. Incidence occurs at a rate of two to three newborns per 10,000 births involving the proliferative and hypertrophic zone of Figure 1: Bilateral genu varum deformity. the physis (epiphysis is normal). Jansen, Schmid and McKusick are the three sub-types with a few reports worldwide [3-9]. Karuna foundation Nepal (KFN) is a non-governmental organization which believes in a world in which each individual, with or without disabilities, has equal access to good quality health care, can lead a dignified life, and can participate as much as possible in community life. KFN approach is entrepreneurial and action oriented, working towards setting up and strengthening existing local health care system, stimulating community participation and responsibility- including health promotion, prevention and rehabilitation through Figure 2: Flat foot. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -
Enzymes and Rna Complexes
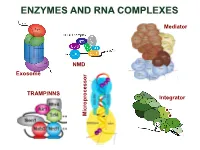
ENZYMES AND RNA COMPLEXES Mediator NMD Exosome NMD TRAMP/NNS Integrator Microprocessor RNA PROCESSING and DECAY machinery: RNases Protein Function Characteristics Exonucleases 5’ 3’ Xrn1 cytoplasmic, mRNA degradation processsive Rat1 nuclear, pre-rRNA, sn/snoRNA, pre-mRNA processing and degradation Rrp17/hNol12 nuclear, pre-rRNA processing Exosome 3’ 5’ multisubunit exo/endo complex subunits organized as in bacterial PNPase Rrp44/Dis3 catalytic subunit Exo/PIN domains, processsive Rrp4, Rrp40 pre-rRNA, sn/snoRNA processing, mRNA degradation Rrp41-43, 45-46 participates in NMD, ARE-dependent, non-stop decay Mtr3, Ski4 Mtr4 nuclear helicase cofactor DEAD box Rrp6 (Rrp47) nuclear exonuclease ( Rrp6 BP, cofactor) RNAse D homolog, processsive Ski2,3,7,8 cytoplasmic exosome cofactors. SKI complex helicase, GTPase Other 3’ 5’ Rex1-4 3’-5’ exonucleases, rRNA, snoRNA, tRNA processing RNase D homolog DXO 3’-5’ exonuclease in addition to decapping mtEXO 3’ 5’ mitochondrial degradosome RNA degradation in yeast Suv3/ Dss1 helicase/ 3’-5’ exonuclease DExH box/ RNase II homolog Deadenylation Ccr4/NOT/Pop2 major deadenylase complex (Ccr, Caf, Pop, Not proteins) Ccr4- Mg2+ dependent endonuclease Pan2p/Pan3 additional deadenylases (poliA tail length) RNase D homolog, poly(A) specific nuclease PARN mammalian deadenylase RNase D homolog, poly(A) specific nuclease Endonucleases RNase III -Rnt1 pre-rRNA, sn/snoRNA processing, mRNA degradation dsRNA specific -Dicer, Drosha siRNA/miRNA biogenesis, functions in RNAi PAZ, RNA BD, RNase III domains Ago2 Slicer -

A Disease-Linked Lncrna Mutation in Rnase MRP Inhibits Ribosome Synthesis
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.29.437572; this version posted March 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A disease-linked lncRNA mutation in RNase MRP inhibits ribosome synthesis Nic Roberston1, Vadim Shchepachev1, David Wright2, Tomasz W. Turowski1, Christos Spanos1, Aleksandra Helwak1, Rose Zamoyska2, David Tollervey1 1 Wellcome Centre for Cell Biology, University of Edinburgh, Edinburgh, UK 2 Ashworth Laboratories, Institute of Immunology and Infection Research, University of Edinburgh, Edinburgh, UK Keywords: protein-RNA interaction; RNA-binding sites; UV crosslinking; mass spectrometry; genetic disease; Cartilage Hair Hypoplasia; ribosome synthesis; T cell activation Running title: RNase MRP defects cause ribosomopathy Highlights: • Mutations in RMRP lncRNA impair pre-rRNA processing and T cell activation • Patient derived fibroblasts show impaired pre-rRNA processing • Cells with the most common disease-linked mutation have specific processing defects • Cytoplasmic ribosomes and intact RNase MRP complexes are also reduced in these cells 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.29.437572; this version posted March 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Mutations in the human RMRP gene cause Cartilage Hair Hypoplasia (CHH), an autosomal recessive disorder characterized by skeletal abnormalities and impaired T cell activation. -

Dwarfism Awareness
LPA Mission Statement LPA is dedicated to improving the quality of life for people with dwarfism throughout their lives, while celebrating with great pride little people’s contribution to social diversity. LPA strives to bring solutions and global For More Information awareness to the prominent issues affecting individuals of Contact LPA short stature and their families. Toll Free…(888) LPA-2001 Direct....(714) 368-3689 Fax…..(707) 721-1896 Dwarfism Check out our website at Awareness www.lpaonline.org A Community Outreach Program 617 Broadway #518 Sponsored by Little People of America Sonoma, CA 95476 LPA is a non-profit tax exempt 501(c)3 organization funded by individual donations. Contact LPA to help. Dwarfism - Facts and Fiction Mythbusters Terminology Bodies come in all shapes and sizes. There are about 400 People with dwarfism are not magical; they do not fly, nor are Preferred terminology is a personal decision, but different types of dwarfism. Each type of dwarfism is they leprechauns, elves, fairies or any other mythological commonly accepted terms are - short stature, different than the other. Many types of dwarfism have creature. They are people - people whose bones happen to dwarfism, little person, dwarf. And we say some medical complications but most people have an grow differently than yours. That is all. "average-height" instead of "normal height". average lifespan, being productive members of society. People with dwarfism do not all know each People with dwarfism are different, yes, but not Eighty percent of people with dwarfism have average- other or look alike, nor are there towns "abnormal". height parents and siblings. -

A Specialized Processing Body That Is Temporally and Asymmetrically Regulated During the Cell Cycle in Saccharomyces Cerevisiae
JCB: ARTICLE A specialized processing body that is temporally and asymmetrically regulated during the cell cycle in Saccharomyces cerevisiae Tina Gill, Jason Aulds, and Mark E. Schmitt Department of Biochemistry and Molecular Biology, State University of New York Upstate Medical University, Syracuse, NY 13210 Nase mitochondrial RNA processing (MRP) is an this spot was asymmetrically found in daughter cells, essential ribonucleoprotein endoribonuclease that where the RNase MRP substrate, CLB2 mRNA, localizes. R functions in the degradation of specifi c mRNAs in- Both the mitotic exit network and fourteen early ana- volved in cell cycle regulation. We have investigated phase release pathways are nonessential but important where this processing event occurs and how it is regu- for the temporal changes in localization. Asymmetric lo- lated. As expected, results demonstrate that RNase MRP calization was found to be dependent on the locasome. is predominantly localized in the nucleolus, where it The evidence suggests that these spots are specialized processes ribosomal RNAs. However, after the initiation processing bodies for the degradation of transcripts that of mitosis, RNase MRP localizes throughout the entire are cell cycle regulated and daughter cell localized. We nucleus and in a single discrete cytoplasmic spot that have called these TAM bodies for temporal asymmetric persists until the completion of telophase. Furthermore, MRP bodies. Introduction RNase mitochondrial RNA processing (MRP) is an essential ri- 27SA preribosomal RNA at the A3 site, forming the 5.8S(s) bonucleoprotein endoribonuclease that cleaves RNA substrates ribosomal RNA (rRNA; Schmitt and Clayton, 1993; Lygerou in a site-specifi c manner and is highly conserved in eukaryotes et al., 1996). -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

A Case Report of Dysosteosclerosis Observed from the Prenatal Period
Clin Pediatr Endocrinol 2010; 19(3), 57-62 Copyright© 2010 by The Japanese Society for Pediatric Endocrinology Case Report A Case Report of Dysosteosclerosis Observed from the Prenatal Period Kisho Kobayashi1, 2, Yusuke Goto1, 2, Hiroaki Kise1, 2, Hiroaki Kanai1, 2, Koji Kodera1, 2, Gen Nishimura3, Kenji Ohyama1, Kanji Sugita1, and Takayuki Komai1, 2 1Department of Pediatrics, University of Yamanashi, Yamanashi, Japan 2Department of Pediatrics, Yamanashi Prefectural Central Hospital, Yamanashi, Japan 3Department of Radiology, Tokyo Metropolitan Children’s Medical Center, Tokyo, Japan Abstract. Dysosteosclerosis is a sclerosing bone dysplasia with skeletal changes resembling those of osteopetrosis. The disorder is associated with dental anomalies and occasionally mental retardation. Because of the rarity and phenotypic diversity of dysosteosclerosis, it remains unsolved whether or not the disorder is heterogeneous. We report here on an affected boy associated with brain calcification and epilepsy with developmental delay. Prenatal ultrasound revealed ventriculomegaly, and brain CT in the neonatal period showed periventricular calcifications. At 13 mo of age, he presented with generalized convulsion with developmental delay. Metaphyseal sclerosis, metaphyseal undermodeling, and oval-shaped vertebral bodies on skeletal survey warranted a diagnosis of dysosteosclerosis. Retrospective review of radiographs as a neonate showed metaphyseal radiolucency, but not metaphyseal sclerosis. Since then, neither the bone changes nor neurological symptom has progressively worsened up to 4 yr of age. Thus, it is thought that the clinical and radiological manifestations of the sclerotic disorder become obvious during infancy. Brain calcification of prenatal onset may be an essential syndromic constituent of the disorder. Key words: dysosteosclerosis, metaphyseal sclerosis, congenital bone disease, periventricular calcification Introduction compression resulting in certain manifestations, such as blindness and facial paralysis. -

Laron Syndrome
Laron syndrome Description Laron syndrome is a rare form of short stature that results from the body's inability to use growth hormone, a substance produced by the brain's pituitary gland that helps promote growth. Affected individuals are close to normal size at birth, but they experience slow growth from early childhood that results in very short stature. If the condition is not treated, adult males typically reach a maximum height of about 4.5 feet; adult females may be just over 4 feet tall. Other features of untreated Laron syndrome include reduced muscle strength and endurance, low blood sugar levels (hypoglycemia) in infancy, small genitals and delayed puberty, hair that is thin and fragile, and dental abnormalities. Many affected individuals have a distinctive facial appearance, including a protruding forehead, a sunken bridge of the nose (saddle nose), and a blue tint to the whites of the eyes (blue sclerae). Affected individuals have short limbs compared to the size of their torso, as well as small hands and feet. Adults with this condition tend to develop obesity. However, the signs and symptoms of Laron syndrome vary, even among affected members of the same family. Studies suggest that people with Laron syndrome have a significantly reduced risk of cancer and type 2 diabetes. Affected individuals appear to develop these common diseases much less frequently than their unaffected relatives, despite having obesity (a risk factor for both cancer and type 2 diabetes). However, people with Laron syndrome do not seem to have an increased lifespan compared with their unaffected relatives. Frequency Laron syndrome is a rare disorder. -

Fibrochondrogenesis
Fibrochondrogenesis Description Fibrochondrogenesis is a very severe disorder of bone growth. Affected infants have a very narrow chest, which prevents the lungs from developing normally. Most infants with this condition are stillborn or die shortly after birth from respiratory failure. However, some affected individuals have lived into childhood. Fibrochondrogenesis is characterized by short stature (dwarfism) and other skeletal abnormalities. Affected individuals have shortened long bones in the arms and legs that are unusually wide at the ends (described as dumbbell-shaped). People with this condition also have a narrow chest with short, wide ribs and a round and prominent abdomen. The bones of the spine (vertebrae) are flattened (platyspondyly) and have a characteristic pinched or pear shape that is noticeable on x-rays. Other skeletal abnormalities associated with fibrochondrogenesis include abnormal curvature of the spine and underdeveloped hip (pelvic) bones. People with fibrochondrogenesis also have distinctive facial features. These include prominent eyes, low-set ears, a small mouth with a long upper lip, and a small chin ( micrognathia). Affected individuals have a relatively flat-appearing midface, particularly a small nose with a flat nasal bridge and nostrils that open to the front rather than downward (anteverted nares). Vision problems, including severe nearsightedness (high myopia) and clouding of the lens of the eye (cataract), are common in those who survive infancy. Most affected individuals also have sensorineural hearing loss, which is caused by abnormalities of the inner ear. Frequency Fibrochondrogenesis appears to be a rare disorder. About 20 affected individuals have been described in the medical literature. Causes Fibrochondrogenesis can result from mutations in the COL11A1 or COL11A2 gene. -

The Rnase MRP and Rnase P Complexes, Will Be Summarised
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/19147 Please be advised that this information was generated on 2021-09-28 and may be subject to change. The human The human RNase MRP complex RNase MRP complex Composition, assembly and role in human disease Hans van Eenennaam Hans van Eenennaam The human RNase MRP complex Composition, assembly and role in human disease Hans van Eenennaam, 2002 The human RNase MRP complex Composition, assembly and role in human disease een wetenschappelijke proeve op het gebied van de Natuurwetenschappen, Wiskunde en Informatica PROEFSCHRIFT ter verkrijging van de graad van doctor aan de Katholieke Universiteit Nijmegen, volgens besluit van het College van Decanen in het openbaar te verdedigen op vrijdag 14 juni 2002 des namiddags om 1:30 uur precies door Hans van Eenennaam Cover illustration Statue of the Dwarf Seneb and his family, painted limestone, height 34 cm, width 22.5 cm, geboren op 3 november 1973 Giza, Tomb of Seneb, Late Fifth - Early Sixth te Middelburg Dynasty. From: The Cairo museum Masterpieces of Egyptian Art, Francesco Tiradritti, Thames & Hudson Ltd, London, 1998 Promotor Prof. Dr. W.J. van Venrooij Co-promotor Dr. G.J.M. Pruijn voor mijn ouders Manuscriptcommissie Prof. Dr. L.B.A. van de Putte Prof. Dr. F.P.J.T. Rutjes Prof. Dr. H.F. Tabak (Universiteit van Amsterdam) ISBN 90-9015696-8 © 2002 by Hans van Eenennaam The research described in this thesis was performed at the Department of Biochemistry, Faculty of Science, University of Nijmegen, the Netherlands. -

Blueprint Genetics Comprehensive Skeletal Dysplasias and Disorders
Comprehensive Skeletal Dysplasias and Disorders Panel Test code: MA3301 Is a 251 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of disorders involving the skeletal system. About Comprehensive Skeletal Dysplasias and Disorders This panel covers a broad spectrum of skeletal disorders including common and rare skeletal dysplasias (eg. achondroplasia, COL2A1 related dysplasias, diastrophic dysplasia, various types of spondylo-metaphyseal dysplasias), various ciliopathies with skeletal involvement (eg. short rib-polydactylies, asphyxiating thoracic dysplasia dysplasias and Ellis-van Creveld syndrome), various subtypes of osteogenesis imperfecta, campomelic dysplasia, slender bone dysplasias, dysplasias with multiple joint dislocations, chondrodysplasia punctata group of disorders, neonatal osteosclerotic dysplasias, osteopetrosis and related disorders, abnormal mineralization group of disorders (eg hypopohosphatasia), osteolysis group of disorders, disorders with disorganized development of skeletal components, overgrowth syndromes with skeletal involvement, craniosynostosis syndromes, dysostoses with predominant craniofacial involvement, dysostoses with predominant vertebral involvement, patellar dysostoses, brachydactylies, some disorders with limb hypoplasia-reduction defects, ectrodactyly with and without other manifestations, polydactyly-syndactyly-triphalangism group of disorders, and disorders with defects in joint formation and synostoses. Availability 4 weeks Gene Set Description