CO2 Separation Using Regenerable Magnesium Solutions Dissolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alkalinity Management in Soilless Substrates and Landscape Architecture Roberto G
Purdue extension HO-242-W Commercial Greenhouse and Nursery Production Purdue Department of Horticulture Alkalinity Management in Soilless Substrates and Landscape Architecture Roberto G. Lopez and Michael V. Mickelbart, Purdue Horticulture and Landscape Architecture www.hort.purdue.edu Ohio State University Horticulture Claudio Pasian, Ohio State University Horticulture and Crop Science and Crop Science www.hcs.osu.edu Purdue Floriculture Greenhouse and nursery growers often assume yellow and pale leaves are signs of nutritional flowers.hort.purdue.edu deficiencies. Sometimes, however, the problem may be due to a lack of specific nutrients in the substrate — in other cases, the nutrients may be present but high substrate pH makes them unavailable to the plant. Growers commonly add sulfuric or other acids to irrigation water to help lower substrate pH or apply iron chelates to quickly “green up” their crops before shipping. Unfortunately, growers often do not have a clear understanding of the underlying causes behind increased substrate pH — water alkalinity, rather than water pH, is typically the source of the problem. The purpose of this publication is to help growers differentiate between high pH and high alkalinity and how to manage alkalinity in soilless substrates. What Is pH? The pH scale measures the concentration of hydrogen ions (H+) in a solution. Examples of solutions are tap water and the water in the substrate inside a pot. The pH scale ranges from 0 to 14. A value of 7.0, which is pure water, is neutral. Values less than 7.0 are called acidic, and values greater than 7 are called basic or alkaline. -

Summary Statistics for Selected Common Ions, Including Dissolved
Common Ions Precambrian Aquifers Summary statistics for selected common ions, Generally, water from the Precambrian aquifers including dissolved solids, calcium, magnesium, is fresh (less than 1,000 mg/L dissolved solids concen- sodium, percent sodium, sodium-adsorption ratio tration). Calcium and bicarbonate generally are dominant among the common ions. Water from the (SAR), potassium, bicarbonate, carbonate, sulfate, Precambrian aquifers has the highest median chloride chloride, fluoride, bromide, iodide, and silica, are concentration, lowest mean and median concentrations presented in table 4. The significance of the various of calcium, magnesium, and bicarbonate, and the common ions is described in table 1. Boxplots are pre- lowest median sulfate (equal to the Deadwood aquifer) sented in figure 16 for each of the common ions, except of the major aquifers. for carbonate. The water type for the various aquifers The water type of the Precambrian aquifers also is discussed in this section. Trilinear diagrams generally is a calcium bicarbonate or a calcium magne- (fig. 17) are presented for each of the aquifers. sium bicarbonate type but also can be a mixed type Changes in water type as ground water flows down- (fig. 17). The original rock mineralogy, degree of gradient are discussed for the Madison, Minnelusa, and metamorphism, and degree of weathering all can Inyan Kara aquifers. contribute to the variations in water type. Two of 56 samples from Precambrian aquifers Specific conductance can be used to estimate the exceed the SMCL of 500 mg/L for dissolved solids. concentration of dissolved solids using the equations Two of 112 samples exceed the SMCL of 250 mg/L for presented in table 5. -

( 12 ) United States Patent
US010682325B2 (12 ) United States Patent ( 10 ) Patent No.: US 10,682,325 B2 Comb et al. (45 ) Date of Patent : * Jun . 16 , 2020 (54 ) COMPOSITIONS AND METHODS FOR THE 3,988,466 A 10/1976 Takagi et al. TREATMENT OF LIVER DISEASES AND 4,496,703 A 1/1985 Steinmetzer 4,871,550 A 10/1989 Millman DISORDERS ASSOCIATED WITH ONE OR 4,898,879 A 2/1990 Madsen et al . BOTH OF HYPERAMMONEMIA OR 4,908,214 A 3/1990 Bobee et al. MUSCLE WASTING 5,028,622 A 7/1991 Plaitakis 5,034,377 A 7/1991 Adibi et al. 5,106,836 A 4/1992 Clemens et al . ( 71) Applicant: AXCELLA HEALTH INC . , 5,229,136 A 7/1993 Mark et al. Cambridge , MA (US ) 5,276,018 A 1/1994 Wilmore 5,348,979 A 9/1994 Nissen et al . (72 ) Inventors : William Comb , Melrose , MA (US ); 5,356,873 A 10/1994 Mark et al . Sean Carroll, Cambridge, MA (US ) ; 5,405,835 A 4/1995 Mendy Raffi Afeyan , Boston , MA (US ) ; 5,438,042 A 8/1995 Schmidl et al. 5,504,072 A 4/1996 Schmidl et al. Michael Hamill, Wellesley , MA (US ) 5,520,948 A 5/1996 Kvamme 5,571,783 A 11/1996 Montagne et al. (73 ) Assignee : AXCELLA HEALTH INC . , 5,576,351 A 11/1996 Yoshimura et al . Cambridge, MA (US ) 5,712,309 A 1/1998 Finnin et al . 5,719,133 A 2/1998 Schmidl et al. Subject to any disclaimer, the term of this 5,719,134 A 2/1998 Schmidl et al. -

The Solubility of Magnesium Carbonate in Aqueous Solutions of Certain
c THE SOLUBILITY OF MAGNESIUM CARBONATE IN AQUEOUS SOLUTIONS OF CERTAIN ELECTROLYTES I BY F. K. CAMERON AND A. SEIDEIJ, P The work herein described was done in connection with and for the most part antedated that upon the solubility of cal- cium carbonate in solutions of other salts, which has already been described.z The solubility of magnesium carbonate is obviously depen- dent upon the partial pressure of the carbon dioxide in the at- mosphere or gas phase in contact with the solution. This has led to the development of this investigation in three parts : I, When the solutions were in contact with ordinary atnios- pheric air containing a small and practically constant percent- age of carbon dioxide. 2. When the atmosphere in contact with the solutions con- tained practically nothing but carbon dioxide and water. 3. When the solutions were in contact with an atmosphere practically free from carbon dioxide. Under these several conditions the ratio of normal carbon- ate to hydrogen carbonate or bicarbonate for any given concen- tration of solution might be expected to vary greatly. Moreover, it was found, in the case of solutions of mag- nesium carbonate, that temperature changes frequently prodiiced relatively large changes in the solubility, and it was therefore necessary to take precautions to prevent variations in this re- spect during the progress of the work. Unless otherwise stated the work described here was done at 23' C. As is well known, solution studies of magnesium carbonate, when other salts are present, have always given great difficulty Published by the permission of the Secretary of Agriculture. -

US3099524.Pdf
3,099,524 United States Patent 0 Patented July 30, 1963 1 2 aluminium and magnesium with a mixture of sodium 3,099,524 hydroxide and sodium canbonate in appropriate amounts. ANTAClD CGMPOSITIONS The aluminium and magnesium salts are preferably the Frederick Grossmith, Pinner, England, assignor to chlorides. Beecham Research Laboratories Limited, Breutford, A further method for preparing the compounds of the Middlesex, England present invention is to suspend a magnesium hydroxo Filed June 28, 1960, Ser. No. 39,245 Claims priority, application Great Britain July 2, 1959 aluminate of the type disclosed in by said co?pending 6 Claims. (Cl. 23—l4) application Serial No. 842,042, ?led September 24, 1959, in a liquor containing bicarbonate ions. A suitable This invention relates to improvements in antacid 10 liquor is a dilute solution of magnesium bicarbonate. compositions and is particularly concerned with new A still further method for preparing the magnesium chemical compounds which have been found to have bicarbonate hydroxoaluminates, which method minimises desirable antacid properties being eminently suitable for the formation of a basic magnesium carbonate impurity, medicinal use for the treatment of gastric hyperacidity. is to react a solution containing aluminium and mag In my co-pending application Serial No. 842,042, ?led 15 nesium salts in appropriate amounts with sodium hy September 24, 1959, now abandoned, I have disclosed droxide at a suitable rate of delivery. An addition of certain novel compounds which are formed by reacting ‘sodium bicarbonate is made at an appropriate stage and aluminium and magnesium salts with caustic allcalis, in a ?nal addition of mixed sodium hydroxide and sodium which the magnesium content, expressed as Mg(OI-I)2, bicarbonate is made to precipitate all the magnesium lies between the approximate limits 2.5% to 75% by 20 without allowing the pH to rise too high. -

Mineral Carbonation and Industrial Uses of Carbon Dioxide 319 7
Chapter 7: Mineral carbonation and industrial uses of carbon dioxide 319 7 Mineral carbonation and industrial uses of carbon dioxide Coordinating Lead Author Marco Mazzotti (Italy and Switzerland) Lead Authors Juan Carlos Abanades (Spain), Rodney Allam (United Kingdom), Klaus S. Lackner (United States), Francis Meunier (France), Edward Rubin (United States), Juan Carlos Sanchez (Venezuela), Katsunori Yogo (Japan), Ron Zevenhoven (Netherlands and Finland) Review Editors Baldur Eliasson (Switzerland), R.T.M. Sutamihardja (Indonesia) 320 IPCC Special Report on Carbon dioxide Capture and Storage Contents EXECUTIVE SUMMARY 321 7.3 Industrial uses of carbon dioxide and its emission reduction potential 330 7.1 Introduction 322 7.3.1 Introduction 330 7.3.2 Present industrial uses of carbon dioxide 332 7.2 Mineral carbonation 322 7.3.3 New processes for CO2 abatement 332 7.2.1 Definitions, system boundaries and motivation 322 7.3.4 Assessment of the mitigation potential of CO2 7.2.2 Chemistry of mineral carbonation 323 utilization 333 7.2.3 Sources of metal oxides 324 7.3.5 Future scope 334 7.2.4 Processing 324 7.2.5 Product handling and disposal 328 References 335 7.2.6 Environmental impact 328 7.2.7 Life Cycle Assessment and costs 329 7.2.8 Future scope 330 Chapter 7: Mineral carbonation and industrial uses of carbon dioxide 321 EXECUTIVE SUMMARY This Chapter describes two rather different options for carbon and recycled using external energy sources. The resulting dioxide (CO2) storage: (i) the fixation of CO2 in the form of carbonated solids must be stored at an environmentally suitable inorganic carbonates, also known as ‘mineral carbonation’ or location. -

Effect of Some Deacidification Agents on Copper-Catalyzed Degrardation
Preservation Research and Testing Series No. 9501 Effect of Some Deacidification Agents on Copper-Catalyzed Degradation of Paper C. J. Shahani and F. H. Hengemihle Preservation Research and Testing office Preservation Directorate Library of Congress Washington, DC November 1995 Effect of Some Deacidifying Agents on Copper-Catalyzed Degradation of Paper Preservation Research and Testing Series No. 9501 November 1995 Page 1 of 12 Library of Congress ♦ Preservation Directorate www.loc.gov/preservation/ Effect of Some Deacidification Agents on Copper-Catalyzed Degradation of Paper Conservation of Historic and Artistic Works on Paper Organized by the Canadian Conservation Institute Ottawa, Canada, October 3-7, 1988 Introduction Paper conservators are all too familiar with artifacts in which iron gall ink or copper in verdigris pigment has caused severe deterioration of the paper base. But even trace concentrations of these metals, which are generally present in paper, contribute significantly to its aging by catalyzing oxidation reactions.1,2,3 Copper and iron contaminants can also cause foxing in paper.4,5 Williams and his co-workers observed that paper samples containing copper acetate, which degraded rapidly when aged at 90˚C and 50˚C relative humidity (RH), were stabilized appreciably after deacidification treatment with magnesium bicarbonate.3 However, deacidification by a Barrow two-step treatment with calcium hydroxide and calcium bicarbonate solutions did not stabilize against the degradative effect of copper acetate to a comparable -

Innovative Technique for Assessment of Groundwater Quality
PINSTECH-171 INNOVATIVE TECHNIQUE FOR ASSESSMENT OF GROUNDWATER QUALITY Niaz Ahmad Manzoor Ahmad M. Ishaq Sajjad RADIATION AND ISOTOPE APPLICATION DIVISION PAKISTAN INSTITUTE OF NUCLEAR SCIENCE AND TECHNOLOGY P.O. NILORE, ISLAMABAD July 2001 CONTENTS ABSTRACT INTRODUCTION GEO-HYDROLOGY RESULTS AND DISCUSSIONS 3.1 Hydro-chemical Data 3.2 Classification of Chemical Analyses 3.2.1 Classical Trilinear Diagrams 3.2.2 Modern Multi-Rectangular Diagrams 3.3 Hydro-Chemical Spatial Variations 3.4 Sodium-Calcium Relationship 3.5 Hydro-geochemical Processes CONCLUSIONS ACKNOWLEDGEMENTS REFERENCES ABSTRACT Groundwater quality of a part of Chaj Doab has been assessed with innovative techniques which are not reported in literature. The concept of triangular coordinates is modified by multi-rectangular ones for the classification of major cations and anions analysed in the ground water. A MuJti-Rectangular Diagram (MRD) has been developed with the combination of rectangular coordinates by virtue of which milli-equivalent per liter percentages (meq/l %) of major cations and anions could be classified into different categories more efficiently as compared to classical trilinear diagrams. Both Piper diagram and MRD are used for the assessment of 259 data sets analysed from ground water of Chaj Doab area, Pakistan. The differentiated ground water types with MRD in the study area are calcium bicarbonate, magnesium bicarbonate, sodium bicarbonate and sodium sulfate. Sodium bicarbonate type emerges as the most abundant type of ground water in the study area. A map showing spatial variation of groundwater quality has been constructed with the help of MRD. This map shows that, in the vicinity of rivers Chenab and Jhelum, calcium bicarbonate type of waters occur while the central area is mainly covered by sodium bicarbonate dominant waters. -

Quantitative Identification of Metastable Magnesium Carbonate
Article pubs.acs.org/est Quantitative Identification of Metastable Magnesium Carbonate Minerals by Solid-State 13C NMR Spectroscopy Jeremy K. Moore,† J. Andrew Surface,† Allison Brenner,† Philip Skemer,§ Mark S. Conradi,‡,† and Sophia E. Hayes*,† † Department of Chemistry, Washington University, One Brookings Drive, Saint Louis, Missouri, 63130, United States ‡ Department of Physics, Washington University, One Brookings Drive, Saint Louis, Missouri, 63130, United States § Department of Earth and Planetary Sciences, Washington University, One Brookings Drive, Saint Louis, Missouri, 63130, United States *S Supporting Information ABSTRACT: In the conversion of CO2 to mineral carbonates for the permanent geosequestration of CO2, there are multiple magnesium carbonate phases that are potential reaction products. Solid-state 13C NMR is demonstrated as an effective tool for distinguishing magnesium carbonate phases and quantitatively characterizing magnesium carbonate mixtures. Several of these mineral phases include magnesite, hydro- magnesite, dypingite, and nesquehonite, which differ in composition by the number of waters of hydration or the number of crystallographic hydroxyl groups. These carbonates often form in mixtures with nearly overlapping 13C NMR resonances which makes their identification and analysis difficult. In this study, these phases have been investigated with solid-state 13C NMR spectroscopy, including both static and magic-angle spinning (MAS) experiments. Static spectra yield chemical shift anisotropy (CSA) lineshapes that are indicative of the site-symmetry variations of the carbon environments. MAS − spectra yield isotropic chemical shifts for each crystallographically inequivalent carbon and spin lattice relaxation times, T1, yield characteristic information that assist in species discrimination. These detailed parameters, and the combination of static and MAS analyses, can aid investigations of mixed carbonates by 13C NMR. -

UNITED STATES PATENT Office 2,275,032 MANUFACTURE of BASIC MAGNESIUM CAR BONATE and EAT NSUATION MATE RAL COMPRS ING SAME Harold W
Patented Mar. 3, 1942 2,275,032 UNITED STATES PATENT office 2,275,032 MANUFACTURE of BASIC MAGNESIUM CAR BONATE AND EAT NSUATION MATE RAL COMPRS ING SAME Harold W. Greider and Roger A. MacArthur, Wyoming, Ohio, assignors to The Philip Carey Manufacturing Company, a corporation of Ohio No Drawing. Application August 16, 1938, Seria No. 225,139 w 8 Claims. (C. 25-156) This invention relates to the manufacture of narily obtained from the dolomite rock during basic magnesium carbonate and heat insulation burning and from the fuel used for the burning. material comprising basic magnesium carbonate. The Solid matter is then Separated from the mix It relates especially to the production of basic ture of insoluble calcium carbonate and soluble magnesium carbonate in a finely-divided, light magnesium bicarbonate by filtration or settling, and bulky state that is particularly adapted for or by combination of these methods of separa employment in the manufacture of heat insula tion. The clarified liquor consists essentially of tion materials. about a 3 per cent. by weight solution of mag Basic magnesium carbonate is widely used in nesium bicarbonate in water. In practice, it is the manufacture of heat insulation materials. 0 not feasible to make Solutions containing more It is believed to have the chemical formula of the bicarbonate, as the carbon dioxide pres 3MgCO3.MgO2H2.3H2O. A heat insulation mate sure and time which are both necessary for the rial that is very widely used at the present time production of higher concentrations of magne and that contains basic magnesium carbonate is sium bicarbonate, are not economically available. -
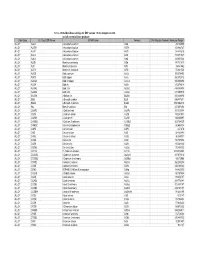
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -
Revision 00 of ANL-EBS-MD-000001, "Environment on Surfaces of Drip Shield & Waste Package Outer Barrier"
DISCLAIMER This contractor document was prepared for the U.S. Departnent of Energy (DOE), but has nor underzone programmatic, policy; or publication review, and is provided for informaon only. The document provides preliminary information that may change based on new information or analysis, and represents a conservative treatment of parameters and assumptions to be used specifically for Total System Performance Assessment analyses. The document is a prelimina,,v lower level contractor document and is not intended for publication or wide distribution. Although this document has under-one tecimical reviews at the contractor organization, it has not undergone a DOE policy review. Therecfore. the .'iews and opinions of authors expressed may not state or reflect those of the DOE. However, in the interest of the rapid transfer of information, we are providing this document for your information per your request. DI5~ 1. QA: OFFIOE OF 0PMLIAN RALIOAC1JVE WASTE MANAGEMENT 1. QA: ANALY818 AWL COVER $MEEfT o(>je a. u Modo Cho*an w PkI C3 Oawmow Modal bxtM O Type of Aidk%at. 0 d~~ w~4t L3 MO%$ uW mm ~M04n4E mw U~tmxmd U.. oAV to oft lfwtd " qm1Icai~ooaaw" kp to 0** MTaV*c ot iSn db MO& ewea; 4, Tfl.: gnl,WfYwlq . it AUVIBOS of ih OLu 8hI uid Wait. PacI Outer Lan1N ANL-ES4A-00m, t 1W 00 m~~~~ ofteDlt,% &jfo odm aatPce u a ;3 ,AO 9.ta wo- Rca--fltY 9.ORA~O COPY~. LAS VEGAS DOCUMENT CONTROL AP-&.lO.i ft. 020m Enclosure 9 OFFICE OF CIVILIAN RADIOACTIVE WASTE MANAGEMENT ANALYSIS/MODEL REVISION RECORD Complete Only Applicable Items 1.