Mutational Analysis and Engineering of the Human Vitamin D Receptor to Bind and Activate in Response to a Novel Small Molecule Ligand
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Left/Right Comparison of Twice-Daily Calcipotriol Ointment and Calcitriol Ointment in Patients with Psoriasis: the Effect on Keratinocyte Subpopulations
Acta Derm Venereol 2004; 84: 195–200 INVESTIGATIVE REPORT A Left/Right Comparison of Twice-Daily Calcipotriol Ointment and Calcitriol Ointment in Patients with Psoriasis: The Effect on Keratinocyte Subpopulations Mannon E.J. FRANSSEN, Gys J. DE JONGH, Piet E.J. VAN ERP and Peter C.M. VAN DE KERKHOF Department of Dermatology, University Medical Centre Nijmegen, The Netherlands Vitamin D3 analogues are a first-line treatment of Calcipotriol (Daivonex1,50mg/g ointment, Leo chronic plaque psoriasis, but so far, comparative clinical Pharmaceutical Products, Denmark) has been investi- studies on calcipotriol and calcitriol ointment are sparse, gated intensively during the last decade, and has proven and in particular no comparative studies are available on to be a valuable tool in the management of chronic cell biological effects of these compounds in vivo. Using plaque psoriasis. A review by Ashcroft et al. (1), based on flow cytometric assessment, we investigated whether these a large number of randomized controlled trials, showed compounds had different effects on the composition and that calcipotriol was at least as effective as potent DNA synthesis of epidermal cell populations responsible topical corticosteroids, 1a,-25-dihydroxycholecalciferol for the psoriatic phenotype. For 8 weeks, 20 patients with (calcitriol), short-contact dithranol, tacalcitol and coal psoriasis vulgaris were treated twice daily with calcipo- tar. Recently, Scott et al. (2) presented an overview of triol and calcitriol ointment in a left/right comparative studies on the use of calcipotriol ointment in the study. Before and after treatment, clinical assessment of management of psoriasis. They reconfirmed the super- target lesions was performed, together with flow cyto- ior efficacy of a twice-daily calcipotriol ointment metric analysis of epidermal subpopulations with respect regimen to the treatments as mentioned above, and to keratin (K) 10, K6, vimentin and DNA distribution. -

Abstract Mahapatra, Debabrata
ABSTRACT MAHAPATRA, DEBABRATA. Vitamin D Receptor and Xenobiotic Interactions: Insights into the diversity and complexity of molecular interactions and their outcomes (Under the direction of Seth W. Kullman). The etiology of a significant number of human diseases including hypertension, cardiovascular disease, diabetes, obesity and cancer are in part associated with exposures to environmental contaminants. Recent trends in toxicological research indicate a growing interest in chemicals capable of disrupting the endocrine system. These chemicals comprise a range of natural and synthetic molecules that interact with nuclear hormone receptors altering signaling pathways regulating adverse effects in multiple organ systems, tissue and cell types. While the interaction of endocrine disrupting xenobiotics with nuclear receptors such as the Estrogen receptor (ER), Thyroid receptor (TR), Glucocorticoid receptor (GR) and Androgen receptor (AR) has been studied in detail, similar research involving xenobiotic interactions with the Vitamin D receptor (VDR) has remained underexplored. This dissertation examines the role of vitamin d receptor as a potential target of xenobiotic induced endocrine disruption and provides new insights into the possible mechanisms underlying the complex molecular interactions between VDR and select VDR agonists and antagonists. Chapter 1 tests the usefulness and importance of orthogonal assays for validation and confirmation of activity profiles of chemicals generated by the qHTS (Quantitative Highthroughput Testing) -

Eldecalcitol Is More Effective for Promoting Osteogenesis Than Alfacalcidol in Cyp27b1-Knockout Mice
bioRxiv preprint doi: https://doi.org/10.1101/349837; this version posted June 18, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Eldecalcitol is more effective for promoting osteogenesis than alfacalcidol in Cyp27b1-knockout Mice Short title: Osteogenic effect of eldecalcitol Yoshihisa Hirota1,2*¶, Kimie Nakagawa2¶, Keigo Isomoto2¶, Toshiyuki Sakaki3, Noboru Kubodera4, Maya Kamao2, Naomi Osakabe5, Yoshitomo Suhara6, Toshio Okano2* 1 Laboratory of Biochemistry, Department of Bioscience and Engineering, College of Systems Engineering and Science, Shibaura Institute of Technology, 307 Fukasaku, Minuma-ku, Saitama 337-8570, Japan 2 Laboratory of Hygienic Sciences, Kobe Pharmaceutical University, 4-19-1 Motoyamakita-machi, Higashinada-ku, Kobe 658-8558, Japan 3 Department of Pharmaceutical Engineering, Faculty of Engineering, Toyama Prefectural University, Kurokawa, Imizu, Toyama 939-0398, Japan 4 International Institute of Active Vitamin D Analogs, 35-6, Sankeidai, Mishima, Shizuoka 411-0017, Japan 5 Food and Nutrition Laboratory, Department of Bioscience and Engineering, College of Systems Engineering and Science, Shibaura Institute of Technology, 307 Fukasaku, Minuma-ku, Saitama 337-8570, Japan 6 Laboratory of Organic Synthesis and Medicinal Chemistry, Department of Bioscience and Engineering, College of Systems Engineering and Science, Shibaura Institute of Technology, 307 Fukasaku, Minuma-ku, Saitama 337-8570, Japan ¶ These authors contributed equally to this work. * Corresponding authors: Yoshihisa Hirota Tel.: +81-48-7201-6037; Fax: +81-48-7201-6011; E-mail: hirotay@ shibaura-it.ac.jp Toshio Okano Tel.: (81) 78-441-7524; Fax: (81) 78-441-7524; E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/349837; this version posted June 18, 2018. -

A New Role for Vitamin D Receptor Activation in Chronic Kidney Disease
A NEW ROLE FOR VITAMIN D RECEPTOR ACTIVATION IN CHRONIC KIDNEY DISEASE José M Valdivielso 1, Jorge Cannata-Andía2, Blai Coll 1 and Elvira Fernández 1 1Laboratorio de Nefrología Experimental, Hospital Universitario Arnau de Vilanova, IRBLLEIDA, REDinREN ISCIII Lleida, Spain. 2Servicio de Metabolismo Óseo y Mineral. Hospital Universitario Central de Asturias. Instituto “Reina Sofía” de Investigación. REDinREN ISCIII. Universidad de Oviedo Running Title: New role of VDRA Correspondence to: Dr. José M Valdivielso, Laboratorio de Nefrología Experimental, IRBLLEIDA Hospital Universitari Arnau de Vilanova Rovira Roure 80 25198 Lleida, Spain. Phone: +34 973003650 Fax: +34 973702213 e-mail: [email protected] Abstract Vitamin D has proven to be much more than a simple ‘calcium hormone’. The fact that the vitamin D receptor has been found in cells not related to mineral metabolism supports that statement. The interest of nephrologists in Vitamin D and its effects beyond mineral metabolism has increased over the last few years, evidencing the importance of this so-called ‘sunshine hormone’. In the present review, we highlight the most recent developments in the traditional use of vitamin D in chronic kidney disease (CKD) patients, namely the control of secondary hyperparathyroidism (sHPT). Furthermore, we also explore the data available regarding the new possible therapeutic uses of vitamin D for the treatment of other complications present in CKD patients, such as vascular calcification, left ventricular hypertrophy or proteinuria. Finally, some still scarce but very promising data regarding a possible role of vitamin D in kidney transplant patients are also reviewed. The available data point to a potential beneficial effect of vitamin D in CKD patients beyond the control of mineral metabolism. -
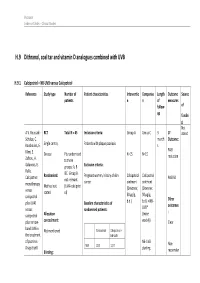
H.9 Dithranol, Coal Tar and Vitamin D Analogues Combined with UVB
Psoriasis Evidence Tables – Clinical Studies H.9 Dithranol, coal tar and vitamin D analogues combined with UVB H.9.1 Calcipotriol + NB-UVB versus Calcipotriol Reference Study type Number of Patient characteristics Interventio Compariso Length Outcome Source patients n n of measures follow- of up fundin g Not A.V. Roussaki- RCT Total N = 45 Inclusion criteria: Group A Group C 3 1º stated Schulze, C. month Outcome: Kouskoukis, E. Single centre, Patients with plaque psoriasis s Klimi, E. PASI Greece Pts randomised N=15 N=15 reduction Zafirou, A. to three Galanous, E. groups: A, B Exclusion criteria: Rallis. &C. Group B Calcipotriol Randomised: Pregnant women, history of skin Calcipotriol Calcipotriol PASI 50 not relevant cancer ointment ointment monotherapy Method not (UVA+calcipotri versus (Dovonex; (Dovonex stated ol) 50 µg/g, 50 µg/g, calcipotriol Other plus UVA1 Baseline characteristics of b.d.) b.d.) + NB- UVB* outcomes versus randomised patients: : calcipotriol Allocation (twice plus narrow- concealment: weekly) Clear band UVB in Calcipotriol Calcipotriol + Not mentioned the treatment NB-UVB of psoriasis. NB-UVB Non- Drugs Exptl. M/F 12/3 12/3 starting Blinding: responder Psoriasis Evidence Tables – Clinical Studies Cl in. Res , dose 80% 31(5/6):169- Not mentioned Age 44.93±6.48 49.53±22.01 MED and 174.2005 inc. by 20% Skin type 0/11/3/1 2/5/6/2 I/II/III/IV every 3 REFID: Washout sessions ROUSSAKISCH period: ULZE2005 90 days if using systemic *Cosmetico therapy, 30 days , 10 lamps if using topicals Helarium B1, 100 W each. -

Is Calcifediol Better Than Cholecalciferol for Vitamin D Supplementation?
Osteoporosis International (2018) 29:1697–1711 https://doi.org/10.1007/s00198-018-4520-y REVIEW Is calcifediol better than cholecalciferol for vitamin D supplementation? J. M. Quesada-Gomez1,2 & R. Bouillon3 Received: 22 February 2018 /Accepted: 28 March 2018 /Published online: 30 April 2018 # International Osteoporosis Foundation and National Osteoporosis Foundation 2018 Abstract Modest and even severe vitamin D deficiency is widely prevalent around the world. There is consensus that a good vitamin D status is necessary for bone and general health. Similarly, a better vitamin D status is essential for optimal efficacy of antiresorptive treatments. Supplementation of food with vitamin D or using vitamin D supplements is the most widely used strategy to improve the vitamin status. Cholecalciferol (vitamin D3) and ergocalciferol (vitamin D2)arethemostwidelyused compounds and the relative use of both products depends on historical or practical reasons. Oral intake of calcifediol (25OHD3) rather than vitamin D itself should also be considered for oral supplementation. We reviewed all publications dealing with a comparison of oral cholecalciferol with oral calcifediol as to define the relative efficacy of both compounds for improving the vitamin D status. First, oral calcifediol results in a more rapid increase in serum 25OHD compared to oral cholecalciferol. Second, oral calcifediol is more potent than cholecalciferol, so that lower dosages are needed. Based on the results of nine RCTs comparing physiologic doses of oral cholecalciferol with oral calcifediol, calcifediol was 3.2-fold more potent than oral chole- calciferol. Indeed, when using dosages ≤ 25 μg/day, serum 25OHD increased by 1.5 ± 0.9 nmol/l for each 1 μgcholecalciferol, whereas this was 4.8 ± 1.2 nmol/l for oral calcifediol. -

Paricalcitol Versus Calcitriol in the Treatment of Secondary Hyperparathyroidism
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Kidney International, Vol. 63 (2003), pp. 1483–1490 Paricalcitol versus calcitriol in the treatment of secondary hyperparathyroidism STUART M. SPRAGUE,FRANCISCO LLACH,MICHAEL AMDAHL,CAROL TACCETTA, and DANIEL BATLLE Division of Nephrology/Hypertension and Department of Medicine, Northwestern University Feinberg School of Medicine, Evanston and Chicago, Illinois; Georgetown University Medical School, Washington, D.C.; and Abbott Laboratories, North Chicago, Illinois Paricalcitol versus calcitriol in the treatment of secondary hyper- Osteitis fibrosa cystica, resulting from poorly con- parathyroidism. trolled secondary hyperparathyroidism, remains a com- Background. Management of secondary hyperparathyroid- mon manifestation of renal osteodystrophy and causes ism has included the use of active vitamin D or vitamin D ana- logs for the suppression of parathyroid hormone (PTH) secre- significant morbidity in patients with chronic renal fail- tion. Although, these agents are effective, therapy is frequently ure [1, 2]. Decreased renal production of calcitriol (1,25 limited by hypercalcemia, hyperphosphatemia, and/or eleva- vitamin D ), hypocalcemia, and hyperphosphatemia are ϫ 3 tions in the calcium-phosphorus (Ca P) product. In clinical the major contributing factors to the development of studies, paricalcitol was shown to be effective at reducing PTH concentrations without causing significant hypercalcemia or secondary hyperparathyroidism [1, 3–5]. Calcitriol is syn- hyperphosphatemia as compared to placebo. A comparative thesized from previtamin D3 by hydroxylation on carbons study was undertaken in order to determine whether paricalci- 25 and 1 in the liver and kidney, respectively [6]. The tol provides a therapeutic advantage to calcitriol. -

Vitamin D and Cancer
WORLD HEALTH ORGANIZATION INTERNATIONAL AGENCY FOR RESEARCH ON CANCER Vitamin D and Cancer IARC 2008 WORLD HEALTH ORGANIZATION INTERNATIONAL AGENCY FOR RESEARCH ON CANCER IARC Working Group Reports Volume 5 Vitamin D and Cancer - i - Vitamin D and Cancer Published by the International Agency for Research on Cancer, 150 Cours Albert Thomas, 69372 Lyon Cedex 08, France © International Agency for Research on Cancer, 2008-11-24 Distributed by WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel: +41 22 791 3264; fax: +41 22 791 4857; email: [email protected]) Publications of the World Health Organization enjoy copyright protection in accordance with the provisions of Protocol 2 of the Universal Copyright Convention. All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the World Health Organization concerning the legal status of any country, territory, city, or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. The mention of specific companies or of certain manufacturer’s products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. The authors alone are responsible for the views expressed in this publication. The International Agency for Research on Cancer welcomes requests for permission to reproduce or translate its publications, in part or in full. -

Lithocholic Acid Is a Vitamin D Receptor Ligand That Acts Preferentially in the Ileum
International Journal of Molecular Sciences Communication Lithocholic Acid Is a Vitamin D Receptor Ligand That Acts Preferentially in the Ileum Michiyasu Ishizawa, Daisuke Akagi and Makoto Makishima * ID Division of Biochemistry, Department of Biomedical Sciences, Nihon University School of Medicine, 30-1 Oyaguchi-kamicho, Itabashi-ku, Tokyo 173-8610, Japan; [email protected] (M.I.); [email protected] (D.A.) * Correspondence: [email protected]; Tel.: +81-3-3972-8111 Received: 26 May 2018; Accepted: 3 July 2018; Published: 6 July 2018 Abstract: The vitamin D receptor (VDR) is a nuclear receptor that mediates the biological action of the active form of vitamin D, 1α,25-dihydroxyvitamin D3 [1,25(OH)2D3], and regulates calcium and bone metabolism. Lithocholic acid (LCA), which is a secondary bile acid produced by intestinal bacteria, acts as an additional physiological VDR ligand. Despite recent progress, however, the physiological function of the LCA−VDR axis remains unclear. In this study, in order to elucidate the differences in VDR action induced by 1,25(OH)2D3 and LCA, we compared their effect on the VDR target gene induction in the intestine of mice. While the oral administration of 1,25(OH)2D3 induced the Cyp24a1 expression effectively in the duodenum and jejunum, the LCA increased target gene expression in the ileum as effectively as 1,25(OH)2D3. 1,25(OH)2D3, but not LCA, increased the expression of the calcium transporter gene Trpv6 in the upper intestine, and increased the plasma calcium levels. Although LCA could induce an ileal Cyp24a1 expression as well as 1,25(OH)2D3, the oral LCA administration was not effective in the VDR target gene induction in the kidney. -

A Randomised Clinical Study of Alfacalcidol and Paricalcitol
PHD THESIS DANISH MEDICAL JOURNAL A Randomised Clinical Study of Alfacalcidol and Paricalcitol Two vitamin D analogs for treatment of secondary hyperparathyroidism in chronic hemodialy- sis patients Ditte Hansen, MD stage 3, and are present in most patients when reaching dialysis. Ninety-six % of the hemodialysis patients (n = 76) in our depart- ment were at the time of screening for participants to the present This review has been accepted as a thesis together with three previously published papers by University of Copnehagen 20th of August 2011 and defended on 8th of study, treated for disturbances in the mineral metabolism. These October 2011 disturbances are associated with alterations in bone morphology, termed renal osteodystrophy and increased risk of skeletal frac- Tutors: Knud Rasmussen and Lisbet Brandi ture. The disturbances in the mineral metabolism are also associ- Official opponents: Klaus Ølgaard, Jens Dam Jensen and Tobias Larsson ated with vascular and other soft tissue calcification, and in turn increased cardiovascular morbidity and mortality. The systemic Correspondence: Department,of Medicine, Roskilde Hospital, Koegevej 7-13, 4000 disorder consisting of mineral disturbances, bone abnormalities Roskilde, Denmark and extraskeletal calcification, is defined as Chronic Kidney Dis- E-mail: [email protected] ease-Mineral and Bone Disorder (CKD-MBD).2 Secondary hyperparathyroidism and renal osteodystrophy Dan Med J 2012;59(2): B4400 When CKD develops 1,25-dihydroxyvitamin D levels decrease.3 This is partly due to decreased availability of the precursor 25- hydroxyvitamin D. The most important reason is the decreased THE THREE ORIGINAL PAPERS ARE 1α-hydroxylation of 25-hydroxyvitamin D in the kidney. -

Strategies for the Synthesis of 19-Nor-Vitamin D Analogs
pharmaceuticals Review Strategies for the Synthesis of 19-nor-Vitamin D Analogs Susana Fernández * and Miguel Ferrero * Departamento de Química Orgánica e Inorgánica, Universidad de Oviedo, 33006 Oviedo, Asturias, Spain * Correspondence: [email protected] (S.F.); [email protected] (M.F.); Tel.: +34-985-102-984 (S.F.); +34-985-105-013 (M.F.) Received: 18 June 2020; Accepted: 21 July 2020; Published: 22 July 2020 Abstract: 1α,25-Dihydroxyvitamin D3 [1α,25-(OH)2-D3], the hormonally active form of vitamin D3, classically regulates bone formation, calcium, and phosphate homeostasis. In addition, this hormone also exerts non-classical effects in a wide variety of target tissues and cell types, such as inhibition of the proliferation and stimulation of the differentiation of normal and malignant cells. However, to produce these actions, supraphysiological doses are required resulting in calcemic effects that limit the use of this natural hormone. During the past 30 years, many structurally modified analogs of the 1α,25-(OH)2-D3 have been synthesized in order to find derivatives that can dissociate the beneficial antiproliferative effects from undesired calcemic effects. Among these candidates, 1α,25-(OH)2-19-nor-D3 analogs have shown promise as good derivatives since they show equal or better activity relative to the parent hormone but with reduced calcemic effects. In this review, we describe the synthetic strategies to obtain the 19-nor-D3 derivatives and briefly describe their physiological activities. Keywords: vitamin D; 19-nor-vitamin D3; 19-nor-vitamin D2; synthesis; modified analogs 1. Introduction The steroid hormone 1α,25-dihydroxyvitamin D3 [1α,25-(OH)2-D3](2, Figure1) is the active form of vitamin D3 (1), which can be synthesized in the skin or obtained from dietary sources [1]. -

A Microbial Metabolite, Lithocholic Acid, Suppresses IFN-Γ and Ahr
bioRxiv preprint doi: https://doi.org/10.1101/491241; this version posted December 9, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. A microbial metabolite, Lithocholic acid, suppresses IFN-g and AhR expression by human cord blood CD4 T cells Anya Nikolai* and Makio Iwashima* *Department of Microbiology and Immunology, Stritch School of Medicine, Loyola University Chicago, Maywood, IL 60153 Corresponding author: Makio Iwashima, Ph.D. Department of Microbiology and Immunology Stritch School of Medicine Loyola University Medical Center Building 115, Rm 270A 2160 S. First Avenue Maywood, IL 60153 Email: [email protected] Phone: 708-216-5816 Fax: 708-216-9574 Declarations of interest: none Highlights • Lithocholic acid suppresses IFNγ production by CD4 T cells. • Lithocholic acid suppresses STAT1 and IRF1 expression by activated CD4 T cells. • Lithocholic acid suppresses AhR in a comparable manner to calcitriol. bioRxiv preprint doi: https://doi.org/10.1101/491241; this version posted December 9, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Vitamin D is a well-known micronutrient that modulates immune responses by epigenetic and transcriptional regulation of target genes, such as inflammatory cytokines. Our group recently demonstrated that the most active form of vitamin D, calcitriol, reduces expression of a transcription factor known as the aryl hydrocarbon receptor (AhR) and inhibits differentiation of a pro-inflammatory T cell subset, Th9. Lithocholic acid (LCA), a secondary bile acid produced by commensal bacteria, is known to bind to and activate the vitamin D receptor (VDR) in a manner comparable to calcitriol.