Roles for Myc in Self-Renewal of Pluripotent Stem Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Meta-Analysis and Inding from Seoul Breast Cancer Study (SEBCS)
The Pharmacogenomics Journal https://doi.org/10.1038/s41397-018-0016-6 ARTICLE Associations between genetic polymorphisms of membrane transporter genes and prognosis after chemotherapy: meta-analysis and finding from Seoul Breast Cancer Study (SEBCS) 1 1 1 1 2 3 4 Ji-Eun Kim ● Jaesung Choi ● JooYong Park ● Chulbum Park ● Se Mi Lee ● Seong Eun Park ● Nan Song ● 5 6 4,7 8 1,4,9 10 Seokang Chung ● Hyuna Sung ● Wonshik Han ● Jong Won Lee ● Sue K. Park ● Mi Kyung Kim ● 4,7 9,11 1,4,9,12 1,4,9 Dong-Young Noh ● Keun-Young Yoo ● Daehee Kang ● Ji-Yeob Choi Received: 7 June 2017 / Revised: 13 October 2017 / Accepted: 4 December 2017 © Macmillan Publishers Limited, part of Springer Nature 2018 Abstract Membrane transporters can be major determinants of the pharmacokinetic profiles of anticancer drugs. The associations between genetic variations of ATP-binding cassette (ABC) and solute carrier (SLC) genes and cancer survival were investigated through a meta-analysis and an association study in the Seoul Breast Cancer Study (SEBCS). Including the SEBCS, the meta-analysis was conducted among 38 studies of genetic variations of transporters on various cancer survivors. 1234567890();,: The population of SEBCS consisted of 1 338 breast cancer patients who had been treated with adjuvant chemotherapy. A total of 7 750 SNPs were selected from 453 ABC and/or SLC genes typed by an Affymetrix 6.0 chip. ABCB1 rs1045642 was associated with poor progression-free survival in a meta-analysis (HR = 1.33, 95% CI: 1.07–1.64). ABCB1, SLC8A1, and SLC12A8 were associated with breast cancer survival in SEBCS (Pgene < 0.05). -

New Products JULY 2013
R&D Systems Tools for Cell Biology Research™ New Products JULY 2013 GMP-grade Recombinant Proteins Contents R&D Systems now offers GMP-grade cytokines and growth factors for research and further manu- facturing applications where current Good Manufacturing Practices (GMP) are required. GMP-grade Recombinant Proteins 2 proteins are manufactured in our ISO-certified facility in compliance with relevant guidelines1 and are produced with extensive documentation at every stage of development from cell culture to final fill and Quantikine® ELISA Kits 3 formulation. GMP-grade Recombinant Human IL-6 and Recombinant Human TNF-a have recently been added to our line of GMP-grade proteins. Additional GMP-grade proteins will be available in the next several months. For an up-to-date product listing or additional information, please visit our website at Luminex® Screening Assays 4 www.RnDSystems.com/GMP. Luminex® Performance Assays 4-5 Features New GMP Proteins ✓ Extensive documentation at every stage of development ProtEin SOURCE Catalog # SIZE Polyclonal Antibodies 6-7 ✓ Documentation of lot-to-lot consistency and traceability Human IL-6 E. coli 206-GMP-010 10 µg Monoclonal Antibodies 7-8 of materials used 206-GMP-050 50 µg ✓ Rigorous quality control using stringent analytical 206-GMP-01M 1 mg processes Biotinylated Antibodies 8 Human TNF-a/ E. coli 210-GMP-010 10 µg ✓ Proven formulations to ensure consistent reconstitution TNFSF1A and results 210-GMP-050 50 µg Antibody Controls 8 210-GMP-01M 1 mg ELISpot Kits & Development Modules 8 30000 60 Fluorokine® Flow Cytometry Kits 8 25000 17348 ) 2 50 20000 Fluorochrome-labeled Antibodies 9 40 8673 DuoSet® ELISA and DuoSet IC ELISA 15000 Development Systems 10 30 Peak Intensity Peak 10000 20 Cell-Based ELISA Assay Kits 10 5000 17565 (Mean RFU x10 Viability Cell 8780 10 Parameter Assay Kits 10 0 0 6000 9000 12000 15000 18000 21000 24000 -3 -2 -1 0 1 10 10 10 10 10 Apoptosis Detection 10 Mass Charge Ratio Recombinant Human TNF-α GMP (ng/mL) MALDI-TOF Analysis of GMP-grade Recombinant Human TNF-a. -

Six3 Overexpression Initiates the Formation of Ectopic Retina
Downloaded from genesdev.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press RESEARCH COMMUNICATION in the developing eye and ventral forebrain. Recently, in Six3 overexpression initiates Drosophila a member of the Six subclass of homeobox the formation of ectopic retina genes, optix, has been isolated, which by sequence com- parison of the homeobox appears to be the ortholog of Felix Loosli, Sylke Winkler, Six3 (Toy et al. 1998). Functional analysis of optix, how- 1 and Joachim Wittbrodt ever, has not yet been reported. In medaka fish (Oryzias latipes) Six3 is expressed in Sonderforschungsbereich (SFB) Junior Group, Institute for Human Genetics, University of Go¨ ttingen, c/o the anterior-most neuroectoderm at gastrula stages and Max-Planck-Institute (MPI) for Biophysical Chemistry, Am later in the developing retina (Loosli et al. 1998). Mosaic Fassberg, 37077 Go¨ttingen, Germany misexpression of mouse Six3 in small clones, in response to injected plasmid DNA, resulted in the formation of The homeobox gene sine oculis (so) is essential for visual ectopic lenses in the region of the otic vesicle, suggesting system formation in Drosophila. A vertebrate member a decisive role for Six3 during vertebrate lens develop- of the so/Six gene family, Six3, is expressed in the devel- ment (Oliver et al. 1996). Considering the expression of oping eye and forebrain. Injection of Six3 RNA into Six3 in the retinal primordia and the essential role of a medaka fish embryos causes ectopic Pax6 and Rx2 ex- Drosophila homolog, so,inDrosophila eye develop- pression in midbrain and cerebellum, resulting in the ment, we investigated a potential role of Six3 in early formation of ectopic retinal primordia. -

Table S1 the Four Gene Sets Derived from Gene Expression Profiles of Escs and Differentiated Cells
Table S1 The four gene sets derived from gene expression profiles of ESCs and differentiated cells Uniform High Uniform Low ES Up ES Down EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol 269261 Rpl12 11354 Abpa 68239 Krt42 15132 Hbb-bh1 67891 Rpl4 11537 Cfd 26380 Esrrb 15126 Hba-x 55949 Eef1b2 11698 Ambn 73703 Dppa2 15111 Hand2 18148 Npm1 11730 Ang3 67374 Jam2 65255 Asb4 67427 Rps20 11731 Ang2 22702 Zfp42 17292 Mesp1 15481 Hspa8 11807 Apoa2 58865 Tdh 19737 Rgs5 100041686 LOC100041686 11814 Apoc3 26388 Ifi202b 225518 Prdm6 11983 Atpif1 11945 Atp4b 11614 Nr0b1 20378 Frzb 19241 Tmsb4x 12007 Azgp1 76815 Calcoco2 12767 Cxcr4 20116 Rps8 12044 Bcl2a1a 219132 D14Ertd668e 103889 Hoxb2 20103 Rps5 12047 Bcl2a1d 381411 Gm1967 17701 Msx1 14694 Gnb2l1 12049 Bcl2l10 20899 Stra8 23796 Aplnr 19941 Rpl26 12096 Bglap1 78625 1700061G19Rik 12627 Cfc1 12070 Ngfrap1 12097 Bglap2 21816 Tgm1 12622 Cer1 19989 Rpl7 12267 C3ar1 67405 Nts 21385 Tbx2 19896 Rpl10a 12279 C9 435337 EG435337 56720 Tdo2 20044 Rps14 12391 Cav3 545913 Zscan4d 16869 Lhx1 19175 Psmb6 12409 Cbr2 244448 Triml1 22253 Unc5c 22627 Ywhae 12477 Ctla4 69134 2200001I15Rik 14174 Fgf3 19951 Rpl32 12523 Cd84 66065 Hsd17b14 16542 Kdr 66152 1110020P15Rik 12524 Cd86 81879 Tcfcp2l1 15122 Hba-a1 66489 Rpl35 12640 Cga 17907 Mylpf 15414 Hoxb6 15519 Hsp90aa1 12642 Ch25h 26424 Nr5a2 210530 Leprel1 66483 Rpl36al 12655 Chi3l3 83560 Tex14 12338 Capn6 27370 Rps26 12796 Camp 17450 Morc1 20671 Sox17 66576 Uqcrh 12869 Cox8b 79455 Pdcl2 20613 Snai1 22154 Tubb5 12959 Cryba4 231821 Centa1 17897 -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -
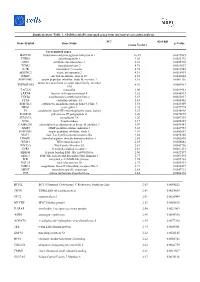
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

Transient Activation of Meox1 Is an Early Component of the Gene
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Access to Research and Communications Annals 1 Transient activation of Meox1 is an early component of the gene 2 regulatory network downstream of Hoxa2. 3 4 Pavel Kirilenko1, Guiyuan He1, Baljinder Mankoo2, Moises Mallo3, Richard Jones4, 5 5 and Nicoletta Bobola1,* 6 7 (1) School of Dentistry, Faculty of Medical and Human Sciences, University of 8 Manchester, Manchester, UK. 9 (2) Randall Division of Cell and Molecular Biophysics, King's College London, UK. 10 (3) Instituto Gulbenkian de Ciência, Oeiras, Portugal. 11 (4) Genetic Medicine, Manchester Academic Health Science Centre, Central 12 Manchester University Hospitals NHS Foundation Trust, Manchester, UK. 13 (5) Present address: Department of Biology, University of York, York, UK. 14 15 Running title: Hoxa2 activates Meox1 expression. 16 Keywords: Meox1, Hoxa2, homeodomain, development, mouse 17 *Words Count: Material and Methods: 344; Introduction, Results and Discussion: 18 3679 19 19 * Author for correspondence at: AV Hill Building The University of Manchester Manchester M13 9PT United Kingdom Phone: (+44) 161 3060642 E-mail: [email protected] 1 Abstract 2 Hox genes encode transcription factors that regulate morphogenesis in all animals 3 with bilateral symmetry. Although Hox genes have been extensively studied, their 4 molecular function is not clear in vertebrates, and only a limited number of genes 5 regulated by Hox transcription factors have been identified. Hoxa2 is required for 6 correct development of the second branchial arch, its major domain of expression. 7 We now show that Meox1 is genetically downstream from Hoxa2 and is a direct 8 target. -

SUPPLEMENTARY MATERIAL Bone Morphogenetic Protein 4 Promotes
www.intjdevbiol.com doi: 10.1387/ijdb.160040mk SUPPLEMENTARY MATERIAL corresponding to: Bone morphogenetic protein 4 promotes craniofacial neural crest induction from human pluripotent stem cells SUMIYO MIMURA, MIKA SUGA, KAORI OKADA, MASAKI KINEHARA, HIROKI NIKAWA and MIHO K. FURUE* *Address correspondence to: Miho Kusuda Furue. Laboratory of Stem Cell Cultures, National Institutes of Biomedical Innovation, Health and Nutrition, 7-6-8, Saito-Asagi, Ibaraki, Osaka 567-0085, Japan. Tel: 81-72-641-9819. Fax: 81-72-641-9812. E-mail: [email protected] Full text for this paper is available at: http://dx.doi.org/10.1387/ijdb.160040mk TABLE S1 PRIMER LIST FOR QRT-PCR Gene forward reverse AP2α AATTTCTCAACCGACAACATT ATCTGTTTTGTAGCCAGGAGC CDX2 CTGGAGCTGGAGAAGGAGTTTC ATTTTAACCTGCCTCTCAGAGAGC DLX1 AGTTTGCAGTTGCAGGCTTT CCCTGCTTCATCAGCTTCTT FOXD3 CAGCGGTTCGGCGGGAGG TGAGTGAGAGGTTGTGGCGGATG GAPDH CAAAGTTGTCATGGATGACC CCATGGAGAAGGCTGGGG MSX1 GGATCAGACTTCGGAGAGTGAACT GCCTTCCCTTTAACCCTCACA NANOG TGAACCTCAGCTACAAACAG TGGTGGTAGGAAGAGTAAAG OCT4 GACAGGGGGAGGGGAGGAGCTAGG CTTCCCTCCAACCAGTTGCCCCAAA PAX3 TTGCAATGGCCTCTCAC AGGGGAGAGCGCGTAATC PAX6 GTCCATCTTTGCTTGGGAAA TAGCCAGGTTGCGAAGAACT p75 TCATCCCTGTCTATTGCTCCA TGTTCTGCTTGCAGCTGTTC SOX9 AATGGAGCAGCGAAATCAAC CAGAGAGATTTAGCACACTGATC SOX10 GACCAGTACCCGCACCTG CGCTTGTCACTTTCGTTCAG Suppl. Fig. S1. Comparison of the gene expression profiles of the ES cells and the cells induced by NC and NC-B condition. Scatter plots compares the normalized expression of every gene on the array (refer to Table S3). The central line -

Supplementary Material
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Page 1 / 45 SUPPLEMENTARY MATERIAL Appendix A1: Neuropsychological protocol. Appendix A2: Description of the four cases at the transitional stage. Table A1: Clinical status and center proportion in each batch. Table A2: Complete output from EdgeR. Table A3: List of the putative target genes. Table A4: Complete output from DIANA-miRPath v.3. Table A5: Comparison of studies investigating miRNAs from brain samples. Figure A1: Stratified nested cross-validation. Figure A2: Expression heatmap of miRNA signature. Figure A3: Bootstrapped ROC AUC scores. Figure A4: ROC AUC scores with 100 different fold splits. Figure A5: Presymptomatic subjects probability scores. Figure A6: Heatmap of the level of enrichment in KEGG pathways. Kmetzsch V, et al. J Neurol Neurosurg Psychiatry 2021; 92:485–493. doi: 10.1136/jnnp-2020-324647 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Appendix A1. Neuropsychological protocol The PREV-DEMALS cognitive evaluation included standardized neuropsychological tests to investigate all cognitive domains, and in particular frontal lobe functions. The scores were provided previously (Bertrand et al., 2018). Briefly, global cognitive efficiency was evaluated by means of Mini-Mental State Examination (MMSE) and Mattis Dementia Rating Scale (MDRS). Frontal executive functions were assessed with Frontal Assessment Battery (FAB), forward and backward digit spans, Trail Making Test part A and B (TMT-A and TMT-B), Wisconsin Card Sorting Test (WCST), and Symbol-Digit Modalities test. -

Transcriptional and Post-Transcriptional Regulation of ATP-Binding Cassette Transporter Expression
Transcriptional and Post-transcriptional Regulation of ATP-binding Cassette Transporter Expression by Aparna Chhibber DISSERTATION Submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Pharmaceutical Sciences and Pbarmacogenomies in the Copyright 2014 by Aparna Chhibber ii Acknowledgements First and foremost, I would like to thank my advisor, Dr. Deanna Kroetz. More than just a research advisor, Deanna has clearly made it a priority to guide her students to become better scientists, and I am grateful for the countless hours she has spent editing papers, developing presentations, discussing research, and so much more. I would not have made it this far without her support and guidance. My thesis committee has provided valuable advice through the years. Dr. Nadav Ahituv in particular has been a source of support from my first year in the graduate program as my academic advisor, qualifying exam committee chair, and finally thesis committee member. Dr. Kathy Giacomini graciously stepped in as a member of my thesis committee in my 3rd year, and Dr. Steven Brenner provided valuable input as thesis committee member in my 2nd year. My labmates over the past five years have been incredible colleagues and friends. Dr. Svetlana Markova first welcomed me into the lab and taught me numerous laboratory techniques, and has always been willing to act as a sounding board. Michael Martin has been my partner-in-crime in the lab from the beginning, and has made my days in lab fly by. Dr. Yingmei Lui has made the lab run smoothly, and has always been willing to jump in to help me at a moment’s notice. -

Supplementary Material Contents
Supplementary Material Contents Immune modulating proteins identified from exosomal samples.....................................................................2 Figure S1: Overlap between exosomal and soluble proteomes.................................................................................... 4 Bacterial strains:..............................................................................................................................................4 Figure S2: Variability between subjects of effects of exosomes on BL21-lux growth.................................................... 5 Figure S3: Early effects of exosomes on growth of BL21 E. coli .................................................................................... 5 Figure S4: Exosomal Lysis............................................................................................................................................ 6 Figure S5: Effect of pH on exosomal action.................................................................................................................. 7 Figure S6: Effect of exosomes on growth of UPEC (pH = 6.5) suspended in exosome-depleted urine supernatant ....... 8 Effective exosomal concentration....................................................................................................................8 Figure S7: Sample constitution for luminometry experiments..................................................................................... 8 Figure S8: Determining effective concentration .........................................................................................................