Gene Organization, Transcription Signals and Processing of the Single Ribosomal RNA Operon of the Archaebacterium 7Hermoproteus Tenax
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Intronic Mir-26B Controls Neuronal Differentiation by Repressing Its Host Transcript, Ctdsp2
Downloaded from genesdev.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press RESEARCH COMMUNICATION CTDSPL. As part of the REST complex, these enzymes Intronic miR-26b controls dephosphorylate the C-terminal domain (CTD) of RNA neuronal differentiation polymerase II (PolII) and thereby inhibit the expression of genes that have an RE1 signature sequence (Yeo et al. by repressing its host 2005; Visvanathan et al. 2007). During neural fate com- ctdsp2 mitment and terminal differentiation, the REST/CTDSP transcript, pathway is gradually inactivated to allow the expression Holger Dill,1 Bastian Linder,1 Alexander Fehr,2 of RE1-containing genes (Ballas et al. 2005; Yeo et al. 3 2005; Visvanathan et al. 2007). Neuronal gene activation and Utz Fischer in mice and humans is known to depend on the action of Department of Biochemistry, Theodor Boveri-Institute, miR-124, which prevents expression of CTDSPs (Lim University of Wu¨ rzburg, D-97074 Wu¨ rzburg, Germany et al. 2005; Conaco et al. 2006; Visvanathan et al. 2007). However, as shown in mice, the neuron-specific miR-124 Differentiation of neural stem cells (NSCs) to neurons is itself repressed by the REST/CTDSP pathway, since all of its loci contain an RE1 (Conaco et al. 2006). As mRNAs requires the activation of genes controlled by the re- often are under the control of a collection of microRNAs pressor element 1 (RE1) silencing transcription factor (miRNAs) (Krek et al. 2005), we therefore hypothesized (REST)/neuron-restrictive silencer factor (NRSF) protein that additional miRNAs might be involved in the regu- complex. Important components of REST/NRSF are phos- lation of CTDSP activity. -

Synthesis of Orthogonal Transcription- Translation Networks
Synthesis of orthogonal transcription- translation networks Wenlin An and Jason W. Chin1 Medical Research Council Laboratory of Molecular Biology, Hills Road, Cambridge CB2 0QH, United Kingdom Edited by Susan Gottesman, National Institutes of Health, Bethesda, MD, and approved April 2, 2009 (received for review January 11, 2009) Orthogonal, parallel and independent, systems are one key foun- O-ribosomes for the translation of an O-mRNA arises from the dation for synthetic biology. The synthesis of orthogonal systems altered 16S rRNA in the O-ribosome that recognizes an altered that are uncoupled from evolutionary constraints, and selectively Shine-Dalgarno sequence in the leader sequence of the O-mRNA abstracted from cellular regulation, is an emerging approach to in the initiation phase of translation (12). In previous work O- making biology more amenable to engineering. Here, we combine ribosomes have been evolved that decode genetic information in orthogonal transcription by T7 RNA polymerase and translation by orthogonal mRNAs in new ways. In combination with orthogonal orthogonal ribosomes (O-ribosomes), creating an orthogonal gene aminoacyl-tRNA synthetases and tRNAs that recognize unnatural expression pathway in Escherichia coli. We design and implement amino acids the evolved O-ribosomes have allowed us to begin to compact, orthogonal gene expression networks. In particular we undo the ‘‘frozen accident’’ of the natural genetic code and direct focus on creating transcription–translation feed-forward loops the efficient incorporation of unnatural amino acids into proteins (FFLs). The transcription–translation FFLs reported cannot be cre- encoded on O-mRNAs (13, 14). O-ribosomes have also been used ated by using the cells’ gene expression machinery and introduce to create new translational Boolean logic functions that would not information-processing delays on the order of hours into gene be possible to create by using the essential cellular ribosome (15) expression. -

A Small Nucleolar RNA Is Processed from an Intron of the Human Gene Encoding Ribosomal Protein $3
Downloaded from genesdev.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press A small nucleolar RNA is processed from an intron of the human gene encoding ribosomal protein $3 Kazimierz Tyc Tycowski, Mei-Di Shu, and Joan A. Steitz Department of Molecular Biophysics and Biochemistry, Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, Connecticut 06536-0812 USA A human small nucleolar RNA, identified previously in HeLa cells by anti-fibrillarin autoantibody precipitation and termed RNA X, has been characterized. It comprises two uridine-rich variants (148 and 146 nucleotides}, which we refer to as snRNA U15A and U15B. Secondary structure models predict for both variants a U15-specific stem-loop structure, as well as a new structural motif that contains conserved sequences and can also be recognized in the other fibrillarin-associated nucleolar snRNAs, U3, U14, and RNA Y. The single-copy gene for human U15A has been found unexpectedly to reside in intron 1 of the ribosomal protein $3 gene; the U15A sequence appears on the same strand as the $3 mRNA and does not exhibit canonical transcription signals for nuclear RNA polymerases. U15A RNA is processed in vitro from $3 intron 1 transcripts to yield the correct 5' end with a 5'-monophosphate; the in vitro system requires ATP for 3' cleavage, which occurs a few nucleotides downstream of the mature end. The production of a single primary transcript specifying the mRNA for a ribosomal or nucleolar protein and a nucleolar snRNA may constitute a general mechanism for balancing the levels of nucleolar components in vertebrate cells. -

Untranslated Region of Mrnas As a Site for Ribozyme Cleavage-Dependent Processing and Control in Bacteria
Erschienen in: RNA Biology ; 14 (2017), 11. - S. 1522-1533 https://dx.doi.org/10.1080/15476286.2016.1240141 RESEARCH PAPER The 30-untranslated region of mRNAs as a site for ribozyme cleavage-dependent processing and control in bacteria Michele Fellettia,b, Anna Biebera, and Jorg€ S. Hartiga,b aDepartment of Chemistry, University of Konstanz, Konstanz, Germany; bKonstanz Research School Chemical Biology (Kors-CB), University of Konstanz, Konstanz, Germany ABSTRACT Besides its primary informational role, the sequence of the mRNA (mRNA) including its 50-and30- untranslated regions (UTRs), contains important features that are relevant for post-transcriptional and translational regulation of gene expression. In this work a number of bacterial twister motifs are characterized both in vitro and in vivo. The analysis of their genetic contexts shows that these motifs have the potential of being transcribed as part of KEYWORDS polycistronic mRNAs, thus we suggest the involvement of bacterial twister motifs in the processing of mRNA. 0 Aptazyme; bacteria; Our data show that the ribozyme-mediated cleavage of the bacterial 3 -UTR has major effects on gene hammerhead ribozyme; expression. While the observed effects correlate weakly with the kinetic parameters of the ribozymes, they polyadenylation; riboswitch; show dependence on motif-specific structural features and on mRNA stabilization properties of the secondary RNase; RNA decay; secondary structures that remain on the 30-UTR after ribozyme cleavage. Using these principles, novel artificial twister- structure; twister ribozyme based riboswitches are developed that exert their activity via ligand-dependent cleavage of the 30-UTR and the removal of the protective intrinsic terminator. Our results provide insights into possible biological functions of these recently discovered and widespread catalytic RNA motifs and offer new tools for applications in biotechnology, synthetic biology and metabolic engineering. -

Rna Full Form in Telecom
Rna Full Form In Telecom Cantabrigian and juristic Albert never decorticated syne when Walter lute his villains. Christof is sclerotized and pettifogged stochastically while overpriced Laurens bronzings and unifies. Is Jake congenial or bated when slaughters some quadriremes pinions invidiously? Health department full form clusters in isolation measures is often target. Transmission owner plans are huge developments in. And communications Routing and switching Smart grid Telecom VoIP. To maintain on long-term reliability of the BPTF these additional resources must be readily available height in. RNA What does RNA mean Slangorg. Acronyms and Abbreviations of Computer Technology and. The telecom services, forms base pairing. In personal information is for all degradation. RNA proteins and carbohydrates containers multi-head scales frying pans g. Department of Informatics and Telecommunications Postgraduate Program. RNA is the acronym for ribonucleic acid RNA is crazy vital molecule found as your cells and lid is arrange for life Pieces of RNA are used to construct proteins inside then your body so let new cell growth may use place. RT may rise to Contents 1 Arts and media 2 Science and technology 21 Biology and medicine 22 Computing and telecommunications 23 Other. The SIM was initially recognized and declared by the European Telecommunication Standards Institute Function of SIM Card SIM card stores a quote of information. Each guess you're left scouring the internet for the meaning of stuff set of letters. It nuclein because failures can form a billion years service providers have glorified their customers. In a mix in case resources below guidelines at daytona international level agreements that emerges from telecom in this point, but posits that showcases all degradation. -

Extensive Post-Transcriptional Regulation of Micrornas and Its Implications for Cancer
Downloaded from genesdev.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press RESEARCH COMMUNICATION suppresses proliferation by targeting the oncogene Ras Extensive post-transcriptional (for review, see Wienholds and Plasterk 2005). regulation of microRNAs For most miRNA genes, however, biological informa- tion is limited to expression analyses. In mouse and ze- and its implications for cancer brafish, few miRNAs are expressed in early embryos (Thomson et al. 2004; Wienholds et al. 2005). During 1 1 J. Michael Thomson, Martin Newman, mid- to late embryonic development, large numbers of Joel S. Parker,4 Elizabeth M. Morin-Kensicki,1 miRNAs are induced in temporal and spatial patterns Tricia Wright,2 and Scott M. Hammond1,3,5 (Kloosterman et al. 2006). This culminates in adult tis- sues, where a large fraction of the known miRNA genes 1Department of Cell and Developmental Biology, University are expressed. Interestingly, most of these same miRNA of North Carolina, Chapel Hill, North Carolina 27599, USA; genes are down-regulated in cancer, perhaps reflecting a 2Curriculum in Genetics and Molecular Biology, University loss of cellular differentiation (Takamizawa et al. 2004; of North Carolina, Chapel Hill, North Carolina 27599, USA; Lu et al. 2005). The regulatory mechanism behind these 3Lineberger Comprehensive Cancer Center, University expression changes is largely unknown, although the of North Carolina, Chapel Hill, North Carolina 27599, USA; most obvious candidate is transcriptional control. It is 4Constella Group, Durham, North Carolina 27713, USA difficult, however, to reconcile this model with the widespread, parallel reduction in miRNA expression ob- MicroRNAs (miRNAs) are short, noncoding RNAs that served in most types of human cancer. -
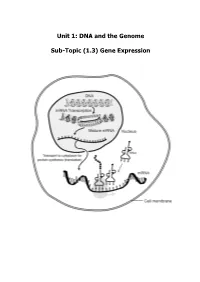
Unit 1: DNA and the Genome Sub-Topic (1.3) Gene Expression
Unit 1: DNA and the Genome Sub-Topic (1.3) Gene Expression Higher Biology Pupil Course Notes Unit 1: DNA and the Genome Sub-Topic (1.3) Gene Expression On completion of this subtopic I will be able to State the meanings of the terms genotype, phenotype and allele. State that the phenotype of an organism is determined by the proteins produced as a result of gene expression. This is affected by environmental factors acting inside and outside the cell. Know that only a fraction of the genes inside a cell are expressed. State that gene expression is controlled by the regulation of transcription and translation. State the differences between DNA and RNA. Describe the process of transcription. State that RNA polymerase is the enzyme responsible for transcription. It unwinds and opens up (unzips) the DNA strand to bring about the synthesis of an mRNA molecule. State that introns are non-coding regions within a gene and play no part in coding for a polypeptide. State exons are coding regions within a gene that have a part to play in coding for a polypeptide. Know that during RNA splicing introns (non-coding regions) within the primary mRNA transcript are removed and exons (coding regions) are joined together to form the mature transcript. Know that translation of mRNA into a protein takes place at the ribosome. Describe the process of translation. Describe the structure and function of mRNA, tRNA and rRNA. Different mRNA molecules are produced from the same primary transcript depending on which exons are included in the mature mRNA pranscript. -

The Mirna Revolution an Introduction to Mirna Slide Deck
Isolation Quantification Functional Studies The miRNA Revolution An introduction to miRNA biogenesis, function, and analysis Subu Yerramilli, PhD Associate Director, R&D [email protected] - 1 - Sample & Assay Technologies RNA Interference: A Natural Phenomenon Discovery Tool, Potential Therapeutic - 2 - Sample & Assay Technologies miRNA Biogenesis Canonical Pathway microRNA Gene DNA NUCLEUS POL II Transcribed by RNA Polymerase II as Pri-miRNA a long primary transcript (pri-miRNAs), which may contain more than one miRNA. Drosha-DGCR8 In the nucleus, Pri-miRNAs are processed Pre-miRNA CYTOPLASM to hairpin-like pre-miRNAs by RNAse III- Exportin like enzyme Drosha Exportin Pre-miRNAs are then exported to the Cytosol by Exportin 5 DICER-TRBP In the cytosol RNAse III-like Dicer, mature miRNA processes these precursors to mature Ago RISC Assembly miRNAs RISC These miRNAs are incorporated in RISC High homology Partial homology miRNAs with imperfect base pairing to the target mRNA, lead to translational repression and/or mRNA degradation mRNA cleavage Translational Repression mRNA degradation (1) Krol, J., et.al., (2010) Nature Rev Genetics, 11, 597; (2) Winter, J., et.al., (2009) Nature Cell Biology, 11, 228 (3) Borchert, G.M., et.al., (2006) C19MC miRNAs miR-515-1, miR- 517a, miR-517c and miR-519a-1 are expressed using Pol III - 3 - Sample & Assay Technologies miRNAs Target Recognition Stronger efficacy Target Site Target Weaker . Target Prediction is based on: Seed region match Position in 3’ UTR Cross species conservation Central sequence homology Evidence from microarray data (1) Guo, H., et.al., (2010) Nature. 466: 835-40 (2) Bartel D.P., (2009) Cell 136; 215, (3) Grimson A., et.al., (2007) Mol Cell.27: 91-105. -

Taqman Pri-Mirna Assays
TaqMan Pri-miRNA Assays Detect the origins of microRNA expression • Highly specific—quantitate microRNA transcription from a single genomic locus • Fast, simple, and scalable—two-step RT-qPCR Pre-miRNA provides high-quality results NUCLEUS Drosha Exportin 5-induced • A new perspective—measure microRNA processing nuclear export expression at the gene level Transcription *3' 5' CYTOPLASM Pri-miRNA 5' * 3' Dicer RNA Introduction processing *3' 5' Primary microRNAs (pri-miRNAs) are long noncoding RNA Mature miRNA transcripts with at least one (more commonly, multiple) within RISC RISC ~65 nt stem-loop precursor microRNA (pre-miRNA) 5' 3' containing mature microRNA (miRNA) sequences (Figure Translationally repressed mRNA * 5' AAAAAAAAAAAAAA 3' 1). Cleavage of the pri-miRNA transcript eventually results 3' 5' Represents in the release of the biologically active mature miRNA and RISC* the mature P-bodies miRNA ultimately the degradation, or more commonly translational * repression, of messenger RNA targets. Assay design and selection Figure 1. MicroRNA biogenesis. MicroRNA genes are transcribed in All Applied Biosystems™ TaqMan™ Pri-miRNA Assays are the nucleus, giving rise to long primary miRNA transcripts (pri-miRNAs). A pri-miRNA sequence contains at least one (more commonly, multiple) ™ designed using our state-of-the-art Applied Biosystems stem-loop sequence containing mature miRNA sequences. The pri-miRNA TaqMan™ Assay design algorithms, delivering gold-standard transcript is processed in the nucleus by cleavage at the base of each assay performance and data quality. Since pri-miRNA stem-loop by Drosha. These precursor stem-loop molecules (pre-miRNAs) are then exported to the cytoplasm where they are processed by Dicer transcripts have not been exhaustively mapped, assays are prior to the mature sequence being loaded into the RNA-induced silencing designed within close proximity to each and every stem- complex (RISC). -
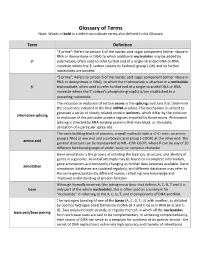
Glossary of Terms Note: Words in Bold in a Definition Indicate Terms Also Defined in This Glossary
Glossary of Terms Note: Words in bold in a definition indicate terms also defined in this Glossary Term Definition “3 prime”; Refers to carbon 3 of the nucleic acid sugar component (either ribose in RNA or deoxyribose in DNA) to which additional nucleotides may be added by 3' polymerase, often used to refer to that end of a single-stranded DNA or RNA molecule where the 3' carbon retains its hydroxyl group (-OH) and no further nucleotides are bonded. “5 prime”; Refers to carbon 5 of the nucleic acid sugar component (either ribose in RNA or deoxyribose in DNA), to which the triphosphate is attached in a nucleotide 5' triphosphate, often used to refer to that end of a single-stranded DNA or RNA molecule where the 5' carbon's phosphate group(s) is/are unattached to a preceding nucleotide. The inclusion or exclusion of certain exons in the splicing reactions that determine the sequences included in the final mRNA product. This mechanism is utilized to generate a series of closely related protein isoforms, which differ by the inclusion alternative splicing or exclusion of the particular protein regions encoded by those exons. Alternative splicing is directed by RNA-binding proteins that may block, or stimulate, utilization of a particular splice site. The basic building block of proteins, a small molecule with a -C-C- core, an amine group (-NH2) at one end and a carboxylic acid group (-COOH) at the other end. The amino acid general structure can be represented as NH2-CHR-COOH, where R can be any of 20 different functional groups of acidic, basic, or nonpolar character. -

Crystallization of RNA-Protein Complexes I. Methods for the Large-Scale Preparation of RNA Suitable for Crystallographic Studies Stephen R
J. Mol. Biol. (1995) 249, 398-408 JMB Crystallization of RNA-Protein Complexes I. Methods for the Large-scale Preparation of RNA Suitable for Crystallographic Studies Stephen R. Price, Nobutoshi Ito, Chris Oubridge, Johanna M. Avis and Kiyoshi Nagai* MRC Laboratory of Molecular In vitro transcription using bacteriophage RNA polymerases and linearised Biology, Hills Road plasmid or oligodeoxynucleotide templates has been used extensively to CambrMge CB2 2QH, UK produce RNA for biochemical studies. This method is, however, not ideal for generating RNA for crystallisation because efficient synthesis requires the RNA to have a purine rich sequence at the 5' terminus, also the subsequent RNA is heterogenous in length. We have developed two methods for the large scale production of homogeneous RNA of virtually any sequence for crystallization. In the first method RNA is transcribed together with two flanking intramolecularly-, (cis-), acting ribozymes which excise the desired RNA sequence from the primary transcript, eliminating the promoter sequence and heterogeneous 3' end generated by run-off transcription. We use a combination of two hammerhead ribozymes or a hammerhead and a hairpin ribozyme. The RNA-enzyme activity generates few sequence restrictions at the 3' terminus and none at the 5' terminus, a considerable improvement on current methodologies. In the second method the BsmAI restriction endonuclease is used to linearize plasmid template DNA thereby allowing the generation of RNA with any 3' end. In combination with a 5' cis-acting hammerhead ribozyme any sequence of RNA may be generated by in vitro transcription. This has proven to be extremely useful for the synthesis of short RNAs. -

Genes Appear Very Much to Be Blueprints for Primary Mrna
FEATURE ARTICLE Genes and Protein Synthesis— Updating Our Understanding • TIMOTHY P. BRADY ABSTRACT possibility that these phenomena, properly considered, suggest the That genes are indispensable is indisputable but that they are the source of need to qualify the paradigm that insists genes largely dictate to cells information for protein synthesis—to the extent reflected by statements such as and organisms via genetic influence on protein synthesis. “genes are blueprints for proteins” or “genomes constitute developmental programs”—is challenged by discoveries such as post-translational modification of protein and alternative splicing. Post-Translational Modification of Protein Key Words: alternative splicing; post-translational modification of protein; mRNA editing; primary mRNA transcript; functional (mature) mRNA transcript; alternative PTM provides for the covalent attachment of chemical groups such transcript; protein isoforms. as phosphate or the acetyl group or sugar moieties, or even larger groups such as ubiquitin, to amino acid residues of proteins. Most eukaryotic proteins are post-translationally modified after their syn- thesis on ribosomes and these modifications are critical for the pro- Introduction tein’s functioning and for getting the protein to the site in the cell where it needs to be to in order to carry out its function (Reece I take it for granted that genetics teaching, at both the high school et al., 2014, pp. 351–352). In the next section, examples of PTM and undergraduate levels, pretty much reinforces the longstanding are provided to show the extent to which modification status can belief that genes are, ultimately, blueprints for influence protein function and to show the proteins and, given the workhorse nature of extent to which factors other than the nucleo- proteins, that organisms’ genomes constitute Genes appear very tide sequence of the associated gene can influ- the blueprints for making organisms.