Electrochemical Reduction of CO2 to Formate on Easily Prepared Carbon-Supported Bi Nanoparticles
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![Frequency and Potential Dependence of Reversible Electrocatalytic Hydrogen Interconversion by [Fefe]-Hydrogenases](https://docslib.b-cdn.net/cover/3536/frequency-and-potential-dependence-of-reversible-electrocatalytic-hydrogen-interconversion-by-fefe-hydrogenases-623536.webp)
Frequency and Potential Dependence of Reversible Electrocatalytic Hydrogen Interconversion by [Fefe]-Hydrogenases
Frequency and potential dependence of reversible electrocatalytic hydrogen interconversion by [FeFe]-hydrogenases Kavita Pandeya,b, Shams T. A. Islama, Thomas Happec, and Fraser A. Armstronga,1 aInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, Oxford OX1 3QR, United Kingdom; bSchool of Solar Energy, Pandit Deendayal Petroleum University, Gandhinagar 382007, Gujarat, India; and cAG Photobiotechnologie Ruhr–Universität Bochum, 44801 Bochum, Germany Edited by Thomas E. Mallouk, The Pennsylvania State University, University Park, PA, and approved March 2, 2017 (received for review December 5, 2016) The kinetics of hydrogen oxidation and evolution by [FeFe]- CrHydA1 contains only the H cluster: in the living cell, it receives hydrogenases have been investigated by electrochemical imped- electrons from photosystem I via a small [2Fe–2S] ferredoxin ance spectroscopy—resolving factors that determine the excep- known as PetF (19). No accessory internal Fe–S clusters are tional activity of these enzymes, and introducing an unusual and present to mediate or store electrons. powerful way of analyzing their catalytic electron transport prop- For enzymes, PFE has focused entirely on net catalytic electron erties. Attached to an electrode, hydrogenases display reversible flow that is observed as direct current (DC) in voltammograms + electrocatalytic behavior close to the 2H /H2 potential, making (2, 3, 10). Use of DC cyclic voltammetry as opposed to single them paradigms for efficiency: the electrocatalytic “exchange” rate potential sweeps alone helps to distinguish rapid, steady-state (measured around zero driving force) is therefore an unusual pa- catalytic activity at different potentials from relatively slow, rameter with theoretical and practical significance. Experiments potential-dependent changes in activity that are revealed as were carried out on two [FeFe]-hydrogenases, CrHydA1 from the hysteresis (10). -

Computational Redox Potential Predictions: Applications to Inorganic and Organic Aqueous Complexes, and Complexes Adsorbed to Mineral Surfaces
Minerals 2014, 4, 345-387; doi:10.3390/min4020345 OPEN ACCESS minerals ISSN 2075-163X www.mdpi.com/journal/minerals Review Computational Redox Potential Predictions: Applications to Inorganic and Organic Aqueous Complexes, and Complexes Adsorbed to Mineral Surfaces Krishnamoorthy Arumugam and Udo Becker * Department of Earth and Environmental Sciences, University of Michigan, 1100 North University Avenue, 2534 C.C. Little, Ann Arbor, MI 48109-1005, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-734-615-6894; Fax: +1-734-763-4690. Received: 11 February 2014; in revised form: 3 April 2014 / Accepted: 13 April 2014 / Published: 24 April 2014 Abstract: Applications of redox processes range over a number of scientific fields. This review article summarizes the theory behind the calculation of redox potentials in solution for species such as organic compounds, inorganic complexes, actinides, battery materials, and mineral surface-bound-species. Different computational approaches to predict and determine redox potentials of electron transitions are discussed along with their respective pros and cons for the prediction of redox potentials. Subsequently, recommendations are made for certain necessary computational settings required for accurate calculation of redox potentials. This article reviews the importance of computational parameters, such as basis sets, density functional theory (DFT) functionals, and relativistic approaches and the role that physicochemical processes play on the shift of redox potentials, such as hydration or spin orbit coupling, and will aid in finding suitable combinations of approaches for different chemical and geochemical applications. Identifying cost-effective and credible computational approaches is essential to benchmark redox potential calculations against experiments. -

"Relationships Between Oxidation-Reduction Potential
Relationships between Oxidation-Reduction Potential, Oxidant, and pH in Drinking Water Cheryl N. James3, Rachel C. Copeland2, and Darren A. Lytle1 1U.S. Environmental Protection Agency, NRMRL, Cincinnati, OH 2University of Cincinnati, Department of Environmental Engineering, Cincinnati, OH 3University of Cincinnati, Department of Chemical Engineering, Cincinnati, OH Abstract –Oxidation and reduction (redox) reactions are very important in drinking water. Oxidation-reduction potential (ORP) measurements reflect the redox state of water. Redox measurements are not widely made by drinking water utilities in part because they are not well understood. The objective of this study was to determine the effect of oxidant type and concentration on the ORP of carbonate buffered water as a function of pH. Oxidants that were studied included: chlorine, monochloramine, potassium permanganate, chlorine dioxide, and oxygen. ORP decreased with increasing pH, regardless of the oxidant type or concentration. ORP increased rapidly with increasing oxidant dosage, particularly at lower concentrations. Differences in the redox potentials of different oxidant systems were also observed. Waters that contained chlorine and chlorine dioxide had the highest ORPs. Tests also revealed that there were inconsistencies with redox electrode measurements. In the standard Zobell reference solution, two identical redox electrodes had nearly the same reading, but in test waters the readings sometimes showed a variation as great as 217.7 mV. Key words: ORP, redox potential, redox chemistry, oxidant, drinking water 1.0 BACKGROUND 1.1 Redox Theory Oxidation-reduction (redox) reactions describe the transfer of electrons between atoms, molecules, or ions. Oxidation and reduction reactions occur simultaneously and together make up an electrochemical couple. -

Ch.14-16 Electrochemistry Redox Reaction
Redox Reaction - the basics ox + red <=> red + ox Ch.14-16 1 2 1 2 Oxidizing Reducing Electrochemistry Agent Agent Redox reactions: involve transfer of electrons from one species to another. Oxidizing agent (oxidant): takes electrons Reducing agent (reductant): gives electrons Redox Reaction - the basics Balance Redox Reactions (Half Reactions) Reduced Oxidized 1. Write down the (two half) reactions. ox1 + red2 <=> red1 + ox2 2. Balance the (half) reactions (Mass and Charge): a. Start with elements other than H and O. Oxidizing Reducing Agent Agent b. Balance O by adding water. c. balance H by adding H+. Redox reactions: involve transfer of electrons from one d. Balancing charge by adding electrons. species to another. (3. Multiply each half reaction to make the number of Oxidizing agent (oxidant): takes electrons electrons equal. Reducing agent (reductant): gives electrons 4. Add the reactions and simplify.) Fe3+ + V2+ → Fe2+ + V3 + Example: Balance the two half reactions and redox Important Redox Titrants and the Reactions reaction equation of the titration of an acidic solution of Na2C2O4 (sodium oxalate, colorless) with KMnO4 (deep purple). Oxidizing Reagents (Oxidants) - 2- 2+ MnO4 (qa ) + C2O4 (qa ) → Mn (qa ) + CO2(g) (1)Potassium Permanganate +qa -qa 2-qa 16H ( ) + 2MnO4 ( ) + 5C2O4 ( ) → − + − 2+ 2+ MnO 4 +8H +5 e → Mn + 4 H2 O 2Mn (qa ) + 8H2O(l) + 10CO2( g) MnO − +4H+ + 3 e − → MnO( s )+ 2 H O Example: Balance 4 2 2 Sn2+ + Fe3+ <=> Sn4+ + Fe2+ − − 2− MnO4 + e→ MnO4 2+ - 3+ 2+ Fe + MnO4 <=> Fe + Mn 1 Important Redox Titrants -

Chemical Redox Agents for Organometallic Chemistry
Chem. Rev. 1996, 96, 877−910 877 Chemical Redox Agents for Organometallic Chemistry Neil G. Connelly*,† and William E. Geiger*,‡ School of Chemistry, University of Bristol, U.K., and Department of Chemistry, University of Vermont, Burlington, Vermont 05405-0125 Received October 3, 1995 (Revised Manuscript Received January 9, 1996) Contents I. Introduction 877 A. Scope of the Review 877 B. Benefits of Redox Agents: Comparison with 878 Electrochemical Methods 1. Advantages of Chemical Redox Agents 878 2. Disadvantages of Chemical Redox Agents 879 C. Potentials in Nonaqueous Solvents 879 D. Reversible vs Irreversible ET Reagents 879 E. Categorization of Reagent Strength 881 II. Oxidants 881 A. Inorganic 881 1. Metal and Metal Complex Oxidants 881 2. Main Group Oxidants 887 B. Organic 891 The authors (Bill Geiger, left; Neil Connelly, right) have been at the forefront of organometallic electrochemistry for more than 20 years and have had 1. Radical Cations 891 a long-standing and fruitful collaboration. 2. Carbocations 893 3. Cyanocarbons and Related Electron-Rich 894 Neil Connelly took his B.Sc. (1966) and Ph.D. (1969, under the direction Compounds of Jon McCleverty) degrees at the University of Sheffield, U.K. Post- 4. Quinones 895 doctoral work at the Universities of Wisconsin (with Lawrence F. Dahl) 5. Other Organic Oxidants 896 and Cambridge (with Brian Johnson and Jack Lewis) was followed by an appointment at the University of Bristol (Lectureship, 1971; D.Sc. degree, III. Reductants 896 1973; Readership 1975). His research interests are centered on synthetic A. Inorganic 896 and structural studies of redox-active organometallic and coordination 1. -

The Reduction Potential of the Couple O3 /O;
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Volume 140. number 2 FEBS LETTERS April 1982 THE REDUCTION POTENTIAL OF THE COUPLE O3 /O;- Consequences for mechanisms of ozone toxicity W. H. KOPPENOL Department of Chemistry, University of Maryland Baltimore County, Catonsville, MD 21228, USA Received 12 January 1982 1. Introduction 0s+HzO+20;-+2H+ (1) The toxicity of ozone, a major constituent of pho. At pH 7 Gibbs energies of formation (AGO;) for Oa, tochemical smog is well documented [ 11. According HZ0 and O;- are 39.1 [ 11, -37.6 and 7.6 kcal/mol*. to the Handbook of Chemistry and Physics [2], the The latter two values follow via AGO’ = -nFM”’ ozone molecule is a powerful two-electron oxidant from the reduction potential at pH 7 of the couples with a standard reduction potential of 2.07 V. The Oz/HzO, 0.82 V and 0,/O;-, -0.33 V [7]. From these products of this reaction are oxygen and water. Since data it is calculated that at this pH reaction (1) is two-electron reactions are normally slow, it might be most unlikely to occur, having a Gibbs energy change relevant to investigate the thermodynamics of two of 13.7 kcal. At alkaline pH, however, the reaction consecutive electron transfers, to determine whether becomes thermodynamically possible, because the other reactive oxygen species could be intermediates. Gibbs energy of formation for water is less negative. Secondary oxidizing agents might be formed [3] as To calculate the reduction potential of the couple suggested by the decrease of glutathione in red blood 0,/O;- one needs to know the Gibbs energy of for- cells upon exposure to ozone. -

Test4 Ch19 Electrochemistry Practice Problems
1 General Chemistry II Jasperse Electrochemistry. Extra Practice Problems Oxidation Numbers p1 Free Energy and Equilibrium p10 Balancing Redox; Electrons Transferred; Oxidizing p2 K Values and Voltage p11 Agents; Reducing Agents Spontaneous Voltaic Electrochemical Cells p4 Nonstandard Concentrations and Cell Potential p11 Cell Potentials p5 Electrolysis p12 Predictable Oxidation and Reduction Strength Patterns p8 Ranking Relative Activity, Based on Observed p9 Answer Key p13 Reactivity or Lack Thereof Key Equations Given for Test: E˚cell=E˚reduction + E˚oxidation ∆G˚ = –96.5nE˚cell (∆G˚ in kJ) Ecell = E˚ – [0.0592/n]log Q log K = nE˚/0.0592 Mol e– = [A • time (sec)/96,500] time (sec)= mol e • 96,500/current (in A) t = (t1/2/0.693) ln (Ao/At) ln (Ao/At) = 0.693•t /t1/2 E = ∆mc2 (m in kg, E in J, c = 3x108 m/s) Oxidation Numbers 1. What is the oxidation number of chromium in the ionic compound ammonium dichromate, (NH4)2Cr2O7? a. +3 d. +6 b. +4 e. +7 c. +5 2. What is the oxidation number of carbon in the ionic compound potassium carbonate, K2CO3? a. +3 d. +6 b. +4 e. +7 c. +5 3. What are the oxidation numbers for nickel, sulfur, and oxygen in Ni2(SO4) 3? a. Ni +3; S +6; O -2 d. Ni +2; S +2; O -2 b. Ni +2; S +4; O -2 e. Ni +2; S +4; O -1 c. Ni +3; S +4; O -2 4. When hydrogen reacts with calcium metal, what are the oxidation numbers of the calcium and hydrogen in the CaH2 product? Ca(s) + H2(g) à CaH2(s) a. -

Field Measurement of Oxidation-Reduction Potential (ORP)
COPY COPY Revision History The top row of this table shows the most recent changes to this controlled document. For previous revision history information, archived versions of this document are maintained by the SESD Document Control Coordinator on the SESD local area network (LAN). History Effective Date SESDPROC-113-R2, Field Measurement of Oxidation-Reduction April 26, 2017 Potential (ORP), replaces SESDPROC-013-R1 General: Corrected any typographical, grammatical, and/or editorial errors. Title Page: Changed the EIB Chief from Danny France to the Field Services Branch Chief John Deatrick, and the Field Quality Manager from Bobby Lewis to Hunter Johnson. Section 2.2: Figure 6 modified for clarity. Section 3.3: Use of overtopping cell described consistent with current practice. SESDPROC-113-R1, Field Measurement of Oxidation-Reduction January 29, 2013 Potential (ORP), replaces SESDPROC-013-R0 SESDPROC-113-R0, Field Measurement of Oxidation-Reduction August 7, 2009 Potential (ORP), Original Issue SESD Operating Procedure Page 2 of 22 SESDPROC-113-R2 Field Measurement of ORP Field Measurement of ORP(113)_AF.R2 Effective Date: April 26, 2017 COPY TABLE OF CONTENTS 1 General Information ............................................................................................................ 4 1.1 Purpose........................................................................................................................... 4 1.2 Scope/Application ........................................................................................................ -

Detection Of
Article Cite This: J. Am. Chem. Soc. 2017, 139, 18552−18557 pubs.acs.org/JACS •− Detection of CO2 in the Electrochemical Reduction of Carbon Dioxide in N,N‑Dimethylformamide by Scanning Electrochemical Microscopy Tianhan Kai, Min Zhou, Zhiyao Duan, Graeme A. Henkelman, and Allen J. Bard* Center for Electrochemistry, Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, United States *S Supporting Information ABSTRACT: The electrocatalytic reduction of CO2 has been studied extensively and produces a number of products. The initial reaction in the CO2 reduction is often taken to be the 1e formation of the radical anion, CO •−. However, the electrochemical detection and •− 2 •− characterization of CO2 is challenging because of the short lifetime of CO2 , which can dimerize and react with proton donors and even mild oxidants. Here, we report the generation •− and quantitative determination of CO2 in N,N-dimethylformamide (DMF) with the tip generation/substrate collection (TG/SC) mode of scanning electrochemical microscopy (SECM). CO2 was reduced at a hemisphere-shaped Hg/Pt ultramicroelectrode (UME) or a fi •− Hg/Au lm UME, which were utilized as the SECM tips. The CO2 produced can either dimerize to form oxalate within the nanogap between SECM tip and substrate or collected at ffi •− SECM substrate (e.g., an Au UME). The collection e ciency (CE) for CO2 depends on the distance (d) between the tip and substrate. The dimerization rate (6.0 × 108 M−1 s−1) and half-life (10 ns) of CO •− can be evaluated by fitting the collection efficiency vs distance curve. The 2 •− dimerized species of CO2 , oxalate, can also be determined quantitatively. -
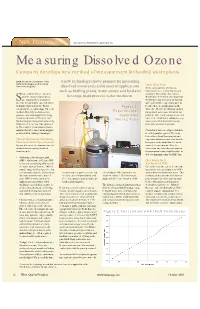
Measuring Dissolved Ozone Company Develops New Method of Measurement for Bottled Water Plants
NEW TECHNOLOGY By Lawrence B. Kilham, EcoSensors, Inc. Measuring Dissolved Ozone Company develops new method of measurement for bottled water plants WQP details the capabilities of the A new technology shows promise for measuring latest technologies on the market Ozone Beta Tests from your suppliers. dissolved ozone and can be used in applications In the early summer of this year, such as bottling plants, water stores and food and instruments were sent out to selected hake a bottle of beer, uncap it, customers for testing (“beta sites”). and the dissolved gases spray beverage plant process water treatment. Results have been better than hoped for. Sout. Sounds like a promising Preliminary top interest is for bottled start for measuring the concentrations water plants where operators want to of dissolved gases in water. That’s Figure 1. be sure there is enough ozone in the essentially the breakthrough. The new water for effective sterilization and yet method efficiently mechanizes this Experimental not so much as to cause a bromate ion gas-water partitioning principle long Apparatus problem. Other applications of current known to chemists as “Henry’s law.” During Tests interest are small water companies, jug Mechanizing gas release from water using water stores and food and beverage Henry’s law is not new. The approach plant process water treatment. is. The result is a new dissolved ozone monitor that overcomes many nagging Consistency and ease of operation have problems with existing technologies. been key points reported. The beta testers have found that analysis and Current Methods for Measuring experimentation is required to find the Dissolved Ozone Concentrations best point in the plant flow stream to By way of review, the common current connect the instrument. -

Basic Concepts in Electrochemistry
1/23/2019 CEE 597T Electrochemical Water and Wastewater Treatment BASIC CONCEPTS IN ELECTROCHEMISTRY What is electrochemistry? ■ Electrochemistry is defined as the branch of chemistry that examines the phenomena resulting from combined chemical and electrical effects. ■ Chemical transformation occurring owing to the external applied electrical current or leading to generation of electrical current is studied in electrochemistry. 1 1/23/2019 Electrochemical Cell An electrochemical cell typically consists of ■ Two electronic conductors (also called electrodes) ■ An ionic conductor (called an electrolyte) ■ the electron conductor used to link the electrodes is often a metal wire, such as copper wiring Types of Cell Galvanic or Voltaic Electrolytic process Processes Reactions in which chemical changes Chemical reactions that result in the occur on the passage of an electrical production of electrical energy. Galvanic current. Electrolytic cells are driven by cells convert chemical potential energy an external source of electrical energy. into electrical energy. A flow of electrons drives non- The energy conversion is achieved by spontaneous (ΔG ≥ 0) redox reactions. spontaneous (ΔG < 0) redox reactions producing a flow of electrons. 2 1/23/2019 Galvanic (Voltaic) Cells The operation of a galvanic (or voltaic) cell is opposite to that of an electrolytic cell. In a galvanic cell, electrical energy is produced by a chemical redox reaction, instead of a chemical reaction being produced by electricity. The classic example of a redox reaction for a galvanic cell is the reaction between aqueous solutions of zinc (Zn) and copper (Cu): In this cell, the zinc is oxidized, and the copper is reduced. Initially, this produces a flow of electrons across a wire connected to the two separate electrode solutions, but as the zinc solution becomes positively charged from losing electrons and the copper solution becomes negatively charged from gaining them, that flow stops. -

Oxidation-Reduction Potential (ORP) for Water Disinfection Monitoring, Control, and Documentation
PUBLICATION 8149 Oxidation-Reduction Potential (ORP) for Water Disinfection Monitoring, Control, and Documentation TREVOR V. SUSLOW, Extension Research Specialist, Department of Vegetable Crops, University of California, Davis Large volumes of water are commonly used during the postharvest handling and UNIVERSITY OF processing of minimally processed fruits and vegetables. Economic considerations CALIFORNIA and wastewater discharge regulations make water recirculation a common practice in Division of Agriculture the industry. Few practices have the capacity of water recirculation to increase the and Natural Resources potential risk of foodborne illness by readily distributing a point source contami- http://anrcatalog.ucdavis.edu nant (one lot, one bin, or even one plant) to noncontaminated produce. Disinfection of water is a critical step in minimizing the potential transmis- sion of pathogens from a water source to produce, among produce within a lot, and between lots over time. Waterborne microorganisms, whether postharvest plant pathogens or agents of human illness, can be rapidly acquired and taken up on plant surfaces. Natural plant surface contours, openings, harvest and trimming wounds, and handling injuries can serve as points of entry for microbes. Microbes within these protected sites are unaffected by common postharvest water treatments such as chlo- rine, chlorine dioxide, ozone, peroxide, peroxyacetic acid, UV irradiation, and other approved treatments at legal rates of application. It is essential, therefore, that the water used for washing, cooling, transporting, postharvest drenching, or other proce- dures be maintained in a condition suitable for the application of these disinfectants. The criteria for the microbial quality of the water become more stringent as product moves from the field to final processing.