Supplementary Materials For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

120421-24Recombschedule FINAL.Xlsx
Friday 20 April 18:00 20:00 REGISTRATION OPENS in Fira Palace 20:00 21:30 WELCOME RECEPTION in CaixaForum (access map) Saturday 21 April 8:00 8:50 REGISTRATION 8:50 9:00 Opening Remarks (Roderic GUIGÓ and Benny CHOR) Session 1. Chair: Roderic GUIGÓ (CRG, Barcelona ES) 9:00 10:00 Richard DURBIN The Wellcome Trust Sanger Institute, Hinxton UK "Computational analysis of population genome sequencing data" 10:00 10:20 44 Yaw-Ling Lin, Charles Ward and Steven Skiena Synthetic Sequence Design for Signal Location Search 10:20 10:40 62 Kai Song, Jie Ren, Zhiyuan Zhai, Xuemei Liu, Minghua Deng and Fengzhu Sun Alignment-Free Sequence Comparison Based on Next Generation Sequencing Reads 10:40 11:00 178 Yang Li, Hong-Mei Li, Paul Burns, Mark Borodovsky, Gene Robinson and Jian Ma TrueSight: Self-training Algorithm for Splice Junction Detection using RNA-seq 11:00 11:30 coffee break Session 2. Chair: Bonnie BERGER (MIT, Cambrige US) 11:30 11:50 139 Son Pham, Dmitry Antipov, Alexander Sirotkin, Glenn Tesler, Pavel Pevzner and Max Alekseyev PATH-SETS: A Novel Approach for Comprehensive Utilization of Mate-Pairs in Genome Assembly 11:50 12:10 171 Yan Huang, Yin Hu and Jinze Liu A Robust Method for Transcript Quantification with RNA-seq Data 12:10 12:30 120 Zhanyong Wang, Farhad Hormozdiari, Wen-Yun Yang, Eran Halperin and Eleazar Eskin CNVeM: Copy Number Variation detection Using Uncertainty of Read Mapping 12:30 12:50 205 Dmitri Pervouchine Evidence for widespread association of mammalian splicing and conserved long range RNA structures 12:50 13:10 169 Melissa Gymrek, David Golan, Saharon Rosset and Yaniv Erlich lobSTR: A Novel Pipeline for Short Tandem Repeats Profiling in Personal Genomes 13:10 13:30 217 Rory Stark Differential oestrogen receptor binding is associated with clinical outcome in breast cancer 13:30 15:00 lunch break Session 3. -

Methodology for Predicting Semantic Annotations of Protein Sequences by Feature Extraction Derived of Statistical Contact Potentials and Continuous Wavelet Transform
Universidad Nacional de Colombia Sede Manizales Master’s Thesis Methodology for predicting semantic annotations of protein sequences by feature extraction derived of statistical contact potentials and continuous wavelet transform Author: Supervisor: Gustavo Alonso Arango Dr. Cesar German Argoty Castellanos Dominguez A thesis submitted in fulfillment of the requirements for the degree of Master’s on Engineering - Industrial Automation in the Department of Electronic, Electric Engineering and Computation Signal Processing and Recognition Group June 2014 Universidad Nacional de Colombia Sede Manizales Tesis de Maestr´ıa Metodolog´ıapara predecir la anotaci´on sem´antica de prote´ınaspor medio de extracci´on de caracter´ısticas derivadas de potenciales de contacto y transformada wavelet continua Autor: Tutor: Gustavo Alonso Arango Dr. Cesar German Argoty Castellanos Dominguez Tesis presentada en cumplimiento a los requerimientos necesarios para obtener el grado de Maestr´ıaen Ingenier´ıaen Automatizaci´onIndustrial en el Departamento de Ingenier´ıaEl´ectrica,Electr´onicay Computaci´on Grupo de Procesamiento Digital de Senales Enero 2014 UNIVERSIDAD NACIONAL DE COLOMBIA Abstract Faculty of Engineering and Architecture Department of Electronic, Electric Engineering and Computation Master’s on Engineering - Industrial Automation Methodology for predicting semantic annotations of protein sequences by feature extraction derived of statistical contact potentials and continuous wavelet transform by Gustavo Alonso Arango Argoty In this thesis, a method to predict semantic annotations of the proteins from its primary structure is proposed. The main contribution of this thesis lies in the implementation of a novel protein feature representation, which makes use of the pairwise statistical contact potentials describing the protein interactions and geometry at the atomic level. -

UC San Diego UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Analysis and applications of conserved sequence patterns in proteins Permalink https://escholarship.org/uc/item/5kh9z3fr Author Ie, Tze Way Eugene Publication Date 2007 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Analysis and Applications of Conserved Sequence Patterns in Proteins A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Computer Science by Tze Way Eugene Ie Committee in charge: Professor Yoav Freund, Chair Professor Sanjoy Dasgupta Professor Charles Elkan Professor Terry Gaasterland Professor Philip Papadopoulos Professor Pavel Pevzner 2007 Copyright Tze Way Eugene Ie, 2007 All rights reserved. The dissertation of Tze Way Eugene Ie is ap- proved, and it is acceptable in quality and form for publication on microfilm: Chair University of California, San Diego 2007 iii DEDICATION This dissertation is dedicated in memory of my beloved father, Ie It Sim (1951–1997). iv TABLE OF CONTENTS Signature Page . iii Dedication . iv Table of Contents . v List of Figures . viii List of Tables . x Acknowledgements . xi Vita, Publications, and Fields of Study . xiii Abstract of the Dissertation . xv 1 Introduction . 1 1.1 Protein Homology Search . 1 1.2 Sequence Comparison Methods . 2 1.3 Statistical Analysis of Protein Motifs . 4 1.4 Motif Finding using Random Projections . 5 1.5 Microbial Gene Finding without Assembly . 7 2 Multi-Class Protein Classification using Adaptive Codes . 9 2.1 Profile-based Protein Classifiers . 14 2.2 Embedding Base Classifiers in Code Space . -

The Pan-NLR'ome of Arabidopsis Thaliana
The pan-NLR’ome of Arabidopsis thaliana Dissertation der Mathematisch-Naturwissenschaftlichen Fakultät der Eberhard Karls Universität Tübingen zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) vorgelegt von Anna-Lena Van de Weyer aus Bad Kissingen Tübingen 2019 Gedruckt mit Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Eberhard Karls Universität Tübingen. Tag der mündlichen Qualifikation: 25.03.2019 Dekan: Prof. Dr. Wolfgang Rosenstiel 1. Berichterstatter: Prof. Dr. Detlef Weigel 2. Berichterstatter: Prof. Dr. Thorsten Nürnberger Acknowledgements I would like to thank my thesis advisors Prof. Detlef Weigel and Prof. Thorsten Nürnberger for not only their continuous support during my PhD studies but also for their critical questions and helpful ideas that shaped and developed my project. Fur- thermore, I would like to thank Dr. Felicity Jones for being part of my Thesis Advisory Committee, for fruitful discussions and for her elaborate questions. I would like to express my sincere gratitude to Dr. Felix Bemm, who worked closely with me during the last years, who continuously challenged and promoted me and with- out whom I would not be where and who I am now. In the mindmap for my acknowl- edgement I wrote down ’Felix: thanks for everything’. That says it all. Further thanks goes to my collaborators Dr. Freddy Monteiro and Dr. Oliver Furzer for their essential contributions to experiments and analyses during all stages of my PhD. Their sheer endless knowledge of NLR biology amplified my curiosity and enthusiasm to learn. Freddy deserves a special thank you for his awesome collaborative spirit and for answering all my NLR related newbie questions. -

Table of Contents
January 10-14, 2015 PLANT & ANIMAL GENOME XXIII Town & Country Hotel THE INTERNATIONAL CONFERENCE San Diego, CA ON THE STATUS OF PLANT & ANIMAL GENOME RESEARCH FINAL PROGRAM, ABSTRACT & EXHIBIT GUIDE Organizing Committee Chairman: Stephen R. Heller, NIST (USA) A PLANT CO-ORGANIZERS A ABSTRACT & WEBSITE Dave Clements, Johns Hopkins University, COORDINATORS USA Victoria Carollo Blake, USDA/ARS/WRRC, Catherine Feuillet, Bayer CropScience, USA USA David Grant, USDA/ARS/CICGR, USA J. Perry Gustafson, University of Missouri, Gerard Lazo, USDA/ARS/WRRC, USA (Retired ), USA Jerome P. Miksche, Emeritus Director, A TRAVEL GRANTS COORDINATOR USDA, Plant Genome Program, USA Douglas Bigwood, Diogenix, USA Graham Moore, John Innes Centre, UK Tom Blake, Montana State University Susan R. Wessler, University of California, (Retired), USA Riverside, USA Rod A. Wing, University of Arizona, USA A SPECIAL DUTY COORDINATORS Hans Cheng, USDA/ARS, USA A ANIMAL CO-ORGANIZERS Max Rothschild, Iowa State University, Ernie Bailey, University of Kentucky, USA USA Daniel Ciobanu, University of Nebraska, USA Kwan-Suk Kim, Chungbuk National University, South Korea Stephen White, USDA/ARS, USA Sponsors and Supporters ORGANIZER Scherago International A USDA, Agricultural Research Service 111 Town Square Place A USDA, National Agricultural Library Suite 1208 USDA, National Institute of Food and Agriculture A Jersey City, NJ 07310 A John Innes Centre Phone: (201) 653-4777 Fax: (201) 653-5705 Cover artwork provided by Applied Biosystems. Originally developed for the company’s -
A Greedy Algorithm for Aligning DNA Sequences
JOURNAL OF COMPUTATIONAL BIOLOGY Volume 7, Numbers 1/2, 2000 Mary Ann Liebert, Inc. Pp. 203–214 A Greedy Algorithm for Aligning DNA Sequences ZHENG ZHANG,1 SCOTT SCHWARTZ,1 LUKAS WAGNER,2 and WEBB MILLER1 ABSTRACT For aligning DNA sequences that differ only by sequencing errors, or by equivalent errors from other sources, a greedy algorithm can be much faster than traditional dynamic pro- gramming approaches and yet produce an alignment that is guaranteed to be theoretically optimal. We introduce a new greedy alignment algorithm with particularly good performance and show that it computes the same alignment as does a certain dynamic programming al- gorithm, while executing over 10 times faster on appropriate data. An implementation of this algorithm is currently used in a program that assembles the UniGene database at the National Center for Biotechnology Information. Key words: sequence alignment, greedy algorithms, dynamic programming. 1. INTRODUCTION et A 5 a1a2 ...aM and B 5 b1b2 ...bN be DNA sequences whose initial portions may be identical Lexcept for sequencing errors. Our goal is to see whether they are, in fact, extremely similar and, if so, how far that similarity extends. For instance, if one but not both of the sequences has had introns spliced out, the similarity might extend only to the end of the rst exon. The fact that only sequencing errors are present, rather than evolutionary changes, means that we should not utilize “afne” gap costs (which penalize each run of consecutive gap columns an additional “gap open” penalty), and this simplies the algorithms. For this discussion, let us assume an error rate of 3%. -

Acknowledgment of Reviewers, 2014
Acknowledgment of Reviewers, 2014 The PNAS editors would like to thank all the individuals who dedicated their considerable time and expertise to the journal by serving as reviewers in 2014. Their generous contribution is deeply appreciated. A Joseph Adams Seungkirl Ahn Hashim Al-Hashimi Luis Amaral Kjersti Aagard-Tillery Michael Adams Tero Ahola Javey Ali Rommie Amaro Duur Aanen Paul Adams Cyrus Aidun Antonios Aliprantis Bruno Amati Jorge Abad Ralf Adams Iannis Aifantis Kari Alitalo Jayakrishna Ambati Alejandro Aballay Lee Adamson Kazuyuki Aihara Rob Alkemade Richard Ambinder Adam Abate John Adelman Judd Aiken Michael Alkire Stanley Ambrose Alireza Abbaspourrad Karen Adelman Elizabeth Ainsworth Robin Allaby Indu Ambudkar Jonathan Abbatt Zach Adelman William Aird Milan P. Allan Chris Amemiya Patrick Abbot Pia Ädelroth Edoardo Airoldi Marc Allard Jan Amend Albert Abbott Sarah Ades Joanna Aizenberg Hunt Allcott Amal Amer Allison Abbott Ilensami Adesida Michael Aizenberg Martin Allday Stefan Ameres Derek Abbott Becky Adkins Myles Akabas Andrew Allen Sebastian Amigorena Larry Abbott Elizabeth Adkins-Regan Ilke Akartuna Charles Allen Mohammed Amin Nicholas Abbott Andy Adler Erol Akcay Dale Allen Ido Amit Paul Abbyad Frederick Adler Mark Akeson David Allen Gabriel Amitai Omar Abdel-Wahab Gregory Adler Christopher Akey Eric Allen Sygal Amitay Yalchin Abdullaev Lynn Adler Ethan Akin Irving Allen Markus Ammann Ikuro Abe Roee Admon Shizuo Akira James Allen David Amodio Jun-ichi Abe Ralph Adolphs Gustav Akk John Allen Angelika Amon Koji Abe Jose Adrio Mikael Akke Karen Allen Christopher Amos Goncalo Abecasis Radoslav Adzic Serap Aksoy Melinda Allen Linda Amos Stephen Abedon Markus Aebi Anastasia Aksyuk Nicola Allen Derk Amsen Markus Abel Toni Aebischer Klaus Aktories Paul Allen Ronald Amundson Moshe Abeles G. -

OE Human Genome Program Contractor-Grantee Workshop IX
Contents Introduction to Contractor-Grantee Workshop IX ....................................................................... l Genomes to Life Program Overview...................................................................................................... 3 Meeting Abstracts ............................................................................................................................................ 5 Sequencing ............................................................................................................................................................... 5 1. The US DOE Joint Genome Institute's High Throughput Production Sequencing Program Susan Lucas, Tijana Glavina, Jamie Jett, Lyle Probst, Andrea Aerts, Nathan Bunker, Sanjay Israni, Astrid Terry, John C. Detter, Sam Pitluck, Heather Kimball, Yunian Lou, Martin Pollard, Anne Olsen, Chris Elkin, Paul Richardson, Dan Rokhsar, Paul Predki, Elbert Branscomb, Trevor Hawkins, and the JGI Sequencing Team.................................................................................................... 5 2. Leveraging Comparative Sequencing Information to Generate a Complete Functional Map of Human Chromosome 19 Lisa Stubbs, Xiaochen Lu, Sha Hammond, Eddie Wehri, Anne Bergmann, Robin Deis, Angela Kolhoff, and Joomyeong Kim ................................................................................. 5 3. The Finishing of Human Chromosomes 19 and 5 Jane Grimwood, Jeremy Schmutz, Mark Dickson, Richard M. Myers, and all members of the Sequencing Group -

Reviewer Acknowledgement 2013 Irene Pala
Pala BMC Bioinformatics 2014, 15:67 http://www.biomedcentral.com/1471-2105/15/67 REVIEWER ACKNOWLEDGEMENT Open Access Reviewer acknowledgement 2013 Irene Pala Contributing reviewers The editors of BMC Bioinformatics would like to thank all our reviewers who have contributed their time to the journal in Volume 14 (2013). Hidenao Abe Dimitra Alexopoulou Cecilia Arighi Japan Germany USA Mohamed Abouelhoda Eivind Almaas Masanori Arita Egypt Norway Japan Jörg Ackermann Uri Alon Martin Aryee Germany Israel USA George Acquaah-Mensah Mohammed Alquraishi Kiyoshi Asai USA USA Japan Alex Adai Fernando Amat Marcella Attimonelli USA USA Italy Pankaj Agarwal Nicola Ancona Raymond Auerbach USA Italy USA Shandar Ahmad Timothy Andersen Frederick Ausubel Japan USA USA Tero Aittokallio Bill Andreopoulos Dmitry Avtonomov Finland Germany USA Martin Akerman Simon Andrews Apostolos Axenopoulos USA UK Greece Mete Akgün Peter Antal Yesim Aydin Son Turkey Hungary Turkey Most M Akhtar Maciek Antoniewicz Vasco Azevedo Italy USA Brazil Tatsuya Akutsu Emilia Apostolova Mohan Babu Japan USA Canada Raul Alcaraz Martinez Sivaram Arabandi Michael Bada Spain USA USA Max Alekseyev Kellie Archer Guy Baele USA USA Belgium Correspondence: [email protected] BioMed Central, 236 Gray’s Inn, Road, London WC1X 8HB, UK © 2014 Pala; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. -

An Introduction to Bioinformatics Algorithms
AN INTRODUCTION TO BIOINFORMATICS ALGORITHMS NEIL C. JONES AND PAVEL A. PEVZNER An Introduction to Bioinformatics Algorithms Sorin Istrail, Pavel Pevzner, and Michael Waterman, editors Computational molecular biology is a new discipline, bringing together com- putational, statistical, experimental, and technological methods, which is energizing and dramatically accelerating the discovery of new technologies and tools for molecular biology. The MIT Press Series on Computational Molecular Biology is intended to provide a unique and effective venue for the rapid publication of monographs, textbooks, edited collections, reference works, and lecture notes of the highest quality. Computational Molecular Biology: An Algorithmic Approach Pavel A. Pevzner, 2000 Computational Methods for Modeling Biochemical Networks James M. Bower and Hamid Bolouri, editors, 2001 Current Topics in Computational Molecular Biology Tao Jiang, Ying Xu, and Michael Q. Zhang, editors, 2002 Gene Regulation and Metabolism: Postgenomic Computation Approaches Julio Collado-Vides, editor, 2002 Microarrays for an Integrative Genomics Isaac S. Kohane, Alvin Kho, and Atul J. Butte, 2002 Kernel Methods in Computational Biology Bernhard Schölkopf, Koji Tsuda, and Jean-Philippe Vert, 2004 An Introduction to Bioinformatics Algorithms Neil C. Jones and Pavel A. Pevzner, 2004 An Introduction to Bioinformatics Algorithms Neil C. Jones Pavel A. Pevzner A Bradford Book The MIT Press Cambridge, Massachusetts London, England © 2004 Massachusetts Institute of Technology All rights reserved. No part of this book may be reproduced in any form by any electronic or mechanical means (including photocopying, recording, or information storage and retrieval) without permission in writing from the publisher. MIT Press books may be purchased at special quantity discounts for business or sales promotional use. -

Comparative Genomics in the Era of Long-Reads an Application on Industrial Yeasts Salazar, A.N
Delft University of Technology Comparative genomics in the era of long-reads An application on industrial yeasts Salazar, A.N. DOI 10.4233/uuid:90594179-e599-4371-ac63-3fa800c53cc9 Publication date 2021 Document Version Final published version Citation (APA) Salazar, A. N. (2021). Comparative genomics in the era of long-reads: An application on industrial yeasts. https://doi.org/10.4233/uuid:90594179-e599-4371-ac63-3fa800c53cc9 Important note To cite this publication, please use the final published version (if applicable). Please check the document version above. Copyright Other than for strictly personal use, it is not permitted to download, forward or distribute the text or part of it, without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license such as Creative Commons. Takedown policy Please contact us and provide details if you believe this document breaches copyrights. We will remove access to the work immediately and investigate your claim. This work is downloaded from Delft University of Technology. For technical reasons the number of authors shown on this cover page is limited to a maximum of 10. Comparative genomics in the era of long-reads An application on industrial yeasts Alex N. Salazar Comparative genomics in the era of long-reads An application on industrial yeasts Dissertation for the purpose of obtaining the degree of doctor at Delft University of Technology by the authority of the Rector Magnificus, Prof.dr.ir. T.H.J.J. van der Hagen chair of the Board of Doctorates to be defended publicly on Friday, 19 February 2021 at 10:00 o’clock by Alex N. -
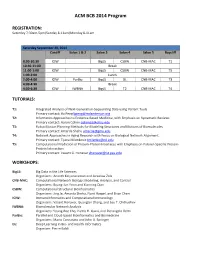
ACM BCB 2014 Program
ACM BCB 2014 Program REGISTRATION: Saturday 7:30am-5pm/Sunday 8-11am/Monday 8-11am Saturday September 20, 2014 Cardiff Salon 1 & 2 Salon 3 Salon 4 Salon 5 Baycliff 8:30-10:30 ICIW BigLS CSBW CNB-MAC T1 10:30-11:00 Break 11:00-1:00 ICIW BigLS CSBW CNB-MAC T5 1:00-2:00 Lunch 2:00-4:00 ICIW ParBio BigLS DL CNB-MAC T3 4:00-4:30 Break 4:30-6:30 ICIW IWBNA BigLS T2 CNB-MAC T4 TUTORIALS: T1: Integrated Analysis of Next-Generation-Sequencing Data using Variant Tools Primary contact: Bo Peng [email protected] T2: Informatics Approaches to Evidence-Based Medicine, with Emphasis on Systematic Reviews Primary contact: Aaron Cohen [email protected] T3: Robot Motion Planning Methods for Modeling Structures and Motions of Biomolecules Primary contact: Amarda Shehu [email protected] T4: Network Approaches in Aging Research with Focus on Biological Network Alignment Primary contact: Tijana Milenkovic [email protected] T5: Computational Prediction of Protein-Protein Interfaces with Emphasis on Partner-Specific Protein- Protein Interaction Primary contact: Vasant G. Honavar [email protected] WORKSHOPS: BigLS: Big Data in the Life Sciences Organizers: Ananth Kalyanaraman and Jaroslaw Zola CNB-MAC: Computational Network Biology: Modeling, Analysis, and Control Organizers: Byung-Jun Yoon and Xiaoning Qian CSBW: Computational Structural Bioinformatics Organizers: Jing Je, Amarda Shehu, Nurit Haspel, and Brian Chen ICIW: Immunoinformatics and Computational Immunology Organizers: Vasant Honavar, Guanglan Zhang, and Lou T. Chitkushev IWBNA: Biomolecular Network Analysis Organizers: Young-Rae Cho, Pietro H. Guzzi, and Pierangelo Veltri ParBio: Parallel and Cloud-based Bioinformatics and Biomedicine Organizers: Mario Cannataro and John A.