Development of Genomic Resources for Nothofagus Species Using Next-Generation Sequencing Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Chapter14.Pdf
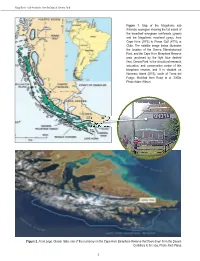
PART I • Omora Park Long-Term Ornithological Research Program THE OMORA PARK LONG-TERM ORNITHOLOGICAL RESEARCH PROGRAM: 1 STUDY SITES AND METHODS RICARDO ROZZI, JAIME E. JIMÉNEZ, FRANCISCA MASSARDO, JUAN CARLOS TORRES-MURA, AND RAJAN RIJAL In January 2000, we initiated a Long-term Ornithological Research Program at Omora Ethnobotanical Park in the world's southernmost forests: the sub-Antarctic forests of the Cape Horn Biosphere Reserve. In this chapter, we first present some key climatic, geographical, and ecological attributes of the Magellanic sub-Antarctic ecoregion compared to subpolar regions of the Northern Hemisphere. We then describe the study sites at Omora Park and other locations on Navarino Island and in the Cape Horn Biosphere Reserve. Finally, we describe the methods, including censuses, and present data for each of the bird species caught in mist nets during the first eleven years (January 2000 to December 2010) of the Omora Park Long-Term Ornithological Research Program. THE MAGELLANIC SUB-ANTARCTIC ECOREGION The contrast between the southwestern end of South America and the subpolar zone of the Northern Hemisphere allows us to more clearly distinguish and appreciate the peculiarities of an ecoregion that until recently remained invisible to the world of science and also for the political administration of Chile. So much so, that this austral region lacked a proper name, and it was generally subsumed under the generic name of Patagonia. For this reason, to distinguish it from Patagonia and from sub-Arctic regions, in the early 2000s we coined the name “Magellanic sub-Antarctic ecoregion” (Rozzi 2002). The Magellanic sub-Antarctic ecoregion extends along the southwestern margin of South America between the Gulf of Penas (47ºS) and Horn Island (56ºS) (Figure 1). -

Bioclimatic and Phytosociological Diagnosis of the Species of the Nothofagus Genus (Nothofagaceae) in South America
International Journal of Geobotanical Research, Vol. nº 1, December 2011, pp. 1-20 Bioclimatic and phytosociological diagnosis of the species of the Nothofagus genus (Nothofagaceae) in South America Javier AMIGO(1) & Manuel A. RODRÍGUEZ-GUITIÁN(2) (1) Laboratorio de Botánica, Facultad de Farmacia, Universidad de Santiago de Compostela (USC). E-15782 Santiago de Com- postela (Galicia, España). Phone: 34-881 814977. E-mail: [email protected] (2) Departamento de Producción Vexetal. Escola Politécnica Superior de Lugo-USC. 27002-Lugo (Galicia, España). E-mail: [email protected] Abstract The Nothofagus genus comprises 10 species recorded in the South American subcontinent. All are important tree species in the ex- tratropical, Mediterranean, temperate and boreal forests of Chile and Argentina. This paper presents a summary of data on the phyto- coenotical behaviour of these species and relates the plant communities to the measurable or inferable thermoclimatic and ombrocli- matic conditions which affect them. Our aim is to update the phytosociological knowledge of the South American temperate forests and to assess their suitability as climatic bioindicators by analysing the behaviour of those species belonging to their most represen- tative genus. Keywords: Argentina, boreal forests, Chile, mediterranean forests, temperate forests. Introduction tually give rise to a temperate territory with rainfall rates as high as those of regions with a Tropical pluvial bio- The South American subcontinent is usually associa- climate; iii. finally, towards the apex of the American ted with a tropical environment because this is in fact the Southern Cone, this temperate territory progressively dominant bioclimatic profile from Panamá to the north of gives way to a strip of land with a Boreal bioclimate. -

Mccaskill Alpine Garden, Lincoln College : a Collection of High
McCaskill Alpine Garden Lincoln College A Collection of High Country Native Plants I/ .. ''11: :. I"" j'i, I Joy M. Talbot Pat V. Prendergast Special Publication No.27 Tussock Grasslands & Mountain Lands Institute. McCaskill Alpine Garden Lincoln College A Collection of High Country Native Plants Text: Joy M. Tai bot Illustration & Design: Pat V. Prendergast ISSN 0110-1781 ISBN O- 908584-21-0 Contents _paQ~ Introduction 2 Native Plants 4 Key to the Tussock Grasses 26 Tussock Grasses 27 Family and Genera Names 32 Glossary 34 Map 36 Index 37 References The following sources were consulted in the compilation of this manual. They are recommended for wider reading. Allan, H. H., 1961: Flora of New Zealand, Volume I. Government Printer, Wellington. Mark, A. F. & Adams, N. M., 1973: New Zealand Alpine Plants. A. H. & A. W. Reed, Wellington. Moore, L.B. & Edgar, E., 1970: Flora of New Zealand, Volume II. Government Printer, Wellington. Poole, A. L. & Adams, N. M., 1980: Trees and Shrubs of New Zealand. Government Printer, Wellington. Wilson, H., 1978: Wild Plants of Mount Cook National Park. Field Guide Publication. Acknowledgement Thanks are due to Dr P. A. Williams, Botany Division, DSIR, Lincoln for checking the text and offering co.nstructive criticism. June 1984 Introduction The garden, named after the founding Director of the Tussock Grasslands and Mountain Lands Institute::', is intended to be educational. From the early 1970s, a small garden plot provided a touch of character to the original Institute building, but it was in 1979 that planning began to really make headway. Land scape students at the College carried out design projects, ideas were selected and developed by Landscape architecture staff in the Department of Horticul ture, Landscape and Parks, and the College approved the proposals. -

Dynamics of Even-Aged Nothofagus Truncata and N. Fusca Stands in North Westland, New Zealand
12 DYNAMICS OF EVEN-AGED NOTHOFAGUS TRUNCATA AND N. FUSCA STANDS IN NORTH WESTLAND, NEW ZEALAND M. C SMALE Forest Research Institute, Private Bag, Rotorua, New Zealand H. VAN OEVEREN Agricultural University, Salverdaplein 11, P.O. Box 9101, 6700 HB, Wageningen, The Netherlands C D. GLEASON* New Zealand Forest Service, P.O. Box 138, Hokitika, New Zealand and M. O. KIMBERLEY Forest Research Institute, Private Bag, Rotorua, New Zealand (Received for publication 30 August, 1985; revision 12 January 1987) ABSTRACT Untended, fully stocked, even-aged stands of Nothofagus truncata (Col.) Ckn. (hard beech) or N. fusca (Hook, f.) Oerst. (red beech) of natural and cultural origin and ranging in age from 20 to 100 years, were sampled using temporary and permanent plots on a range of sites in North Westland, South Island, New Zealand. Changes in stand parameters with age were quantified in order to assess growth of these stands, and thus gain some insight into their silvicultural potential. Stands of each species followed a similar pattern of growth, with rapid early height and basal area increment. Mean top height reached a maximum of c. 27 m by age 100 years. Basal area reached an equilibrium of c. 41 m2/ha in N. truncata and 46 m2/ha in N. fusca as early as age 30 years. Nothofagus truncata stands had, on average, a somewhat lower mean diameter at any given age than N. fusca stands, and maintained higher stockings. Both species attained similar maximum volume of c. 460 m3/ha at age 100 years. Keywords: even-aged stands; stand dynamics; growth; Nothofagus truncata; Nothofagus fusca. -

Regeneration of Subalpine Nothofagus Pumilio in Northwestern Patagonia Argentina
University of Montana ScholarWorks at University of Montana Graduate Student Theses, Dissertations, & Professional Papers Graduate School 2000 Regeneration of subalpine Nothofagus pumilio in northwestern Patagonia Argentina Christopher A. Wall The University of Montana Follow this and additional works at: https://scholarworks.umt.edu/etd Let us know how access to this document benefits ou.y Recommended Citation Wall, Christopher A., "Regeneration of subalpine Nothofagus pumilio in northwestern Patagonia Argentina" (2000). Graduate Student Theses, Dissertations, & Professional Papers. 6805. https://scholarworks.umt.edu/etd/6805 This Thesis is brought to you for free and open access by the Graduate School at ScholarWorks at University of Montana. It has been accepted for inclusion in Graduate Student Theses, Dissertations, & Professional Papers by an authorized administrator of ScholarWorks at University of Montana. For more information, please contact [email protected]. Maureen and Mike MANSFIELD LIBRARY The University of JV IO N T A N A Permission is granted by the author to reproduce this material in its entirety, provided that this material is used for scholarly purposes and is properly cited in published works and reports. ** Please check "Yes" or and provide signature ** Yes, I grant permission k No, I do not grant permission Author’s Signature Date S - P ^ 6 O O Any copying for commercial purposes or financial gain may be undertaken only with the author’s explicit consent. Regeneration of SubalpineNothofagus pumilio in Northwestern Patagonia, Argentina By Christopher A. Wall B.A. Vassar College, 1991 Presented in partial fulfillment of the requirements for the Degree of Master of Science The University of Montana 2000 Approved By Committee Co-Chairs Dean of the Graduate School S'^Zo ' Date UMI Number: EP37606 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. -

Impact of Extreme Weather Events on Aboveground Net Primary Productivity and Sheep Production in the Magellan Region, Southernmost Chilean Patagonia
geosciences Article Impact of Extreme Weather Events on Aboveground Net Primary Productivity and Sheep Production in the Magellan Region, Southernmost Chilean Patagonia Pamela Soto-Rogel 1,* , Juan-Carlos Aravena 2, Wolfgang Jens-Henrik Meier 1, Pamela Gross 3, Claudio Pérez 4, Álvaro González-Reyes 5 and Jussi Griessinger 1 1 Institute of Geography, Friedrich–Alexander-University of Erlangen–Nürnberg, 91054 Erlangen, Germany; [email protected] (W.J.-H.M.); [email protected] (J.G.) 2 Centro de Investigación Gaia Antártica, Universidad de Magallanes, Punta Arenas 6200000, Chile; [email protected] 3 Servicio Agrícola y Ganadero (SAG), Punta Arenas 6200000, Chile; [email protected] 4 Private Consultant, Punta Arenas 6200000, Chile; [email protected] 5 Hémera Centro de Observación de la Tierra, Escuela de Ingeniería Forestal, Facultad de Ciencias, Universidad Mayor, Camino La Pirámide 5750, Huechuraba, Santiago 8580745, Chile; [email protected] * Correspondence: [email protected] Received: 28 June 2020; Accepted: 13 August 2020; Published: 16 August 2020 Abstract: Spatio-temporal patterns of climatic variability have effects on the environmental conditions of a given land territory and consequently determine the evolution of its productive activities. One of the most direct ways to evaluate this relationship is to measure the condition of the vegetation cover and land-use information. In southernmost South America there is a limited number of long-term studies on these matters, an incomplete network of weather stations and almost no database on ecosystems productivity. In the present work, we characterized the climate variability of the Magellan Region, southernmost Chilean Patagonia, for the last 34 years, studying key variables associated with one of its main economic sectors, sheep production, and evaluating the effect of extreme weather events on ecosystem productivity and sheep production. -

Chile: a Journey to the End of the World in Search of Temperate Rainforest Giants
Eliot Barden Kew Diploma Course 53 July 2017 Chile: A Journey to the end of the world in search of Temperate Rainforest Giants Valdivian Rainforest at Alerce Andino Author May 2017 1 Eliot Barden Kew Diploma Course 53 July 2017 Table of Contents 1. Title Page 2. Contents 3. Table of Figures/Introduction 4. Introduction Continued 5. Introduction Continued 6. Aims 7. Aims Continued / Itinerary 8. Itinerary Continued / Objective / the Santiago Metropolitan Park 9. The Santiago Metropolitan Park Continued 10. The Santiago Metropolitan Park Continued 11. Jardín Botánico Chagual / Jardin Botanico Nacional, Viña del Mar 12. Jardin Botanico Nacional Viña del Mar Continued 13. Jardin Botanico Nacional Viña del Mar Continued 14. Jardin Botanico Nacional Viña del Mar Continued / La Campana National Park 15. La Campana National Park Continued / Huilo Huilo Biological Reserve Valdivian Temperate Rainforest 16. Huilo Huilo Biological Reserve Valdivian Temperate Rainforest Continued 17. Huilo Huilo Biological Reserve Valdivian Temperate Rainforest Continued 18. Huilo Huilo Biological Reserve Valdivian Temperate Rainforest Continued / Volcano Osorno 19. Volcano Osorno Continued / Vicente Perez Rosales National Park 20. Vicente Perez Rosales National Park Continued / Alerce Andino National Park 21. Alerce Andino National Park Continued 22. Francisco Coloane Marine Park 23. Francisco Coloane Marine Park Continued 24. Francisco Coloane Marine Park Continued / Outcomes 25. Expenditure / Thank you 2 Eliot Barden Kew Diploma Course 53 July 2017 Table of Figures Figure 1.) Valdivian Temperate Rainforest Alerce Andino [Photograph; Author] May (2017) Figure 2. Map of National parks of Chile Figure 3. Map of Chile Figure 4. Santiago Metropolitan Park [Photograph; Author] May (2017) Figure 5. -

Bosque Pehuén Park's Flora: a Contribution to the Knowledge of the Andean Montane Forests in the Araucanía Region, Chile Author(S): Daniela Mellado-Mansilla, Iván A
Bosque Pehuén Park's Flora: A Contribution to the Knowledge of the Andean Montane Forests in the Araucanía Region, Chile Author(s): Daniela Mellado-Mansilla, Iván A. Díaz, Javier Godoy-Güinao, Gabriel Ortega-Solís and Ricardo Moreno-Gonzalez Source: Natural Areas Journal, 38(4):298-311. Published By: Natural Areas Association https://doi.org/10.3375/043.038.0410 URL: http://www.bioone.org/doi/full/10.3375/043.038.0410 BioOne (www.bioone.org) is a nonprofit, online aggregation of core research in the biological, ecological, and environmental sciences. BioOne provides a sustainable online platform for over 170 journals and books published by nonprofit societies, associations, museums, institutions, and presses. Your use of this PDF, the BioOne Web site, and all posted and associated content indicates your acceptance of BioOne’s Terms of Use, available at www.bioone.org/page/terms_of_use. Usage of BioOne content is strictly limited to personal, educational, and non-commercial use. Commercial inquiries or rights and permissions requests should be directed to the individual publisher as copyright holder. BioOne sees sustainable scholarly publishing as an inherently collaborative enterprise connecting authors, nonprofit publishers, academic institutions, research libraries, and research funders in the common goal of maximizing access to critical research. R E S E A R C H A R T I C L E ABSTRACT: In Chile, most protected areas are located in the southern Andes, in mountainous land- scapes at mid or high altitudes. Despite the increasing proportion of protected areas, few have detailed inventories of their biodiversity. This information is essential to define threats and develop long-term • integrated conservation programs to face the effects of global change. -

Nutrient Cycling in Nothofagus Pumilio Forests Along an Altitudinal Gradient in Tierra Del Fuego, Argentina Jorge L
Forest Ecology and Management 217 (2005) 80–94 www.elsevier.com/locate/foreco Nutrient cycling in Nothofagus pumilio forests along an altitudinal gradient in Tierra del Fuego, Argentina Jorge L. Frangi a, Marcelo D. Barrera a, Laura L. Richter a, Ariel E. Lugo b,* a Laboratorio de Investigacio´n de Sistemas Ecolo´gicos y Ambientales (LISEA), Universidad Nacional de La Plata. CC 31, 1900 La Plata, Argentina b International Institute of Tropical Forestry, USDA Forest Service, P.O. Box 25000, Rı´o Piedras, PR 00928-5000, USA Received 17 November 2004; received in revised form 20 May 2005; accepted 23 May 2005 Abstract Nothofagus pumilio (lenga) forests form monocultures from sea level to timberline in Tierra de Fuego, Argentina. Past studies suggested that the life form change from erect forest to krummholz had advantages to forest function. Aboveground net primary productivity (NPP) and organic matter production per unit leaf area and growing season day were higher in krummholz than in adjacent short erect forests at lower elevation. We compared tall erect, short erect, and krummholz lenga stands in terms of the concentration, accumulation, fluxes, turnover, and use-efficiency of nutrients (N, P, K, Ca, and Mg) along an elevation gradient (220–640 m) in Tierra del Fuego (Valle de Andorra, 54890S, 68820W). With few exceptions, patterns of decreasing values of nutrient concentration, nutrient stock, nutrient flux, and nutrient turnover reversed at the krummholz, which had higher values of these parameters than an adjacent short erect forest at lower elevation. Nutrient cycles accelerated at the krummholz but nutrient use-efficiency of organic matter production and nutrient return to the forest floor decreased. -

Survival and Growth of Nothofagus Pumilio Seedlings Under Several Microenvironments After Variable Retention Harvesting in Southern Patagonian Forests Guillermo J
Survival and growth of Nothofagus pumilio seedlings under several microenvironments after variable retention harvesting in southern Patagonian forests Guillermo J. Martínez Pastur, Rosina Soler Esteban, Juan Cellini, María V. Lencinas, Pablo L. Peri, Mark G. Neyland To cite this version: Guillermo J. Martínez Pastur, Rosina Soler Esteban, Juan Cellini, María V. Lencinas, Pablo L. Peri, et al.. Survival and growth of Nothofagus pumilio seedlings under several microenvironments after variable retention harvesting in southern Patagonian forests. Annals of Forest Science, Springer Nature (since 2011)/EDP Science (until 2010), 2014, 71 (3), pp.349 - 362. 10.1007/s13595-013-0343-3. hal- 01101533 HAL Id: hal-01101533 https://hal.archives-ouvertes.fr/hal-01101533 Submitted on 8 Jan 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Annals of Forest Science (2014) 71:349–362 DOI 10.1007/s13595-013-0343-3 ORIGINAL PAPER Survival and growth of Nothofagus pumilio seedlings under several microenvironments after variable retention harvesting in southern Patagonian forests Guillermo J. Martínez Pastur & Rosina Soler Esteban & Juan M. Cellini & María V. Lencinas & Pablo L. Peri & Mark G. Neyland Received: 30 July 2013 /Accepted: 30 October 2013 /Published online: 22 November 2013 # INRA and Springer-Verlag France 2013 Abstract forest structure, microclimate, soil properties, and nutrient & Context Variable retention prescriptions for Nothofagus availability. -

Shoot Development and Dieback in Progenies of Nothofagus Obliqua Javier G
Shoot development and dieback in progenies of Nothofagus obliqua Javier G. Puntieri, Javier E. Grosfeld, Marina Stecconi, Cecilia Brion, María Marta Azpilicueta, Leonardo Gallo, Daniel Barthélémy To cite this version: Javier G. Puntieri, Javier E. Grosfeld, Marina Stecconi, Cecilia Brion, María Marta Azpilicueta, et al.. Shoot development and dieback in progenies of Nothofagus obliqua. Annals of Forest Science, Springer Nature (since 2011)/EDP Science (until 2010), 2007, 64 (8), pp.839-844. 10.1051/forest:2007068. hal-00259241 HAL Id: hal-00259241 https://hal.archives-ouvertes.fr/hal-00259241 Submitted on 30 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Copyright Ann. For. Sci. 64 (2007) 839–844 Available online at: c INRA, EDP Sciences, 2007 www.afs-journal.org DOI: 10.1051/forest:2007068 Original article Shoot development and dieback in progenies of Nothofagus obliqua Javier Puntieria,b*,JavierGrosfelda,b,MarinaStecconib, Cecilia Briona, María Marta Azpilicuetac, Leonardo Galloc,DanielBarthel´ emy´ d a Departamento de Botánica, Universidad Nacional del Comahue, Quintral 1250, 8400, Bariloche, Argentina b Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina c Laboratorio de Genética Forestal, Instituto Nacional de Tecnología Agropecuaria, EEA Bariloche, Argentina d INRA, Unité Mixte de Recherche CIRAD-CNRS-INRA-IRD-Université Montpellier 2, “ botAnique et bioinforMatique de l’Architecture des Plantes ” (AMAP), UMR T51 (CIRAD), UMR 5120 (CNRS), UMR 931 (INRA), M123 (IRD), UM27 (UMII) TA A-51/PS2, Blvd. -

Commonality and Variability in the Structural Attributes of Moist Temperate Old-Growth Forests: a Global Review ⇑ Sabina Burrascano A, William S
Forest Ecology and Management 291 (2013) 458–479 Contents lists available at SciVerse ScienceDirect Forest Ecology and Management journal homepage: www.elsevier.com/locate/foreco Review Commonality and variability in the structural attributes of moist temperate old-growth forests: A global review ⇑ Sabina Burrascano a, William S. Keeton b, Francesco M. Sabatini a, , Carlo Blasi a a Department of Environmental Biology, Sapienza University of Rome, Rome, Italy b Rubenstein School of Environment and Natural Resources, University of Vermont, Burlington, VT 05405, USA article info abstract Article history: Temperate forests have been fundamentally altered by land use and other stressors globally; these have Received 3 August 2012 reduced the abundance of primary and old-growth forests in particular. Despite many regional studies, Received in revised form 15 October 2012 the literature lacks a global synthesis of temperate old-growth structural characteristics. In this study Accepted 18 November 2012 we compare literature derived data on mature and old-growth moist temperate forests with the aim of: (i) exploring global commonalities; (ii) investigating sources of variability among systems; and (iii) highlighting data gaps and research needs. We compiled a dataset of 147 records from 93 papers, and Keywords: analyzed a set of structural indicators: basal area, stem density, large living trees, live aboveground bio- Literature search mass, quadratic mean diameter, and coarse woody debris volume. These indicators were contrasted Forest dynamics Sustainable forest management between mature and old-growth age classes at a global level and across continents and broad forest Carbon sequestration types, testing for significance through Monte-Carlo permutation procedure. We also related structural Biodiversity indicators to age, climatic and geographical descriptors.