Immunopathology of Organ Transplantation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clinical Pathology, Immunopathology and Advanced Vaccine Technology in Bovine Theileriosis: a Review
pathogens Review Clinical Pathology, Immunopathology and Advanced Vaccine Technology in Bovine Theileriosis: A Review Onyinyechukwu Ada Agina 1,2,* , Mohd Rosly Shaari 3, Nur Mahiza Md Isa 1, Mokrish Ajat 4, Mohd Zamri-Saad 5 and Hazilawati Hamzah 1,* 1 Department of Veterinary Pathology and Microbiology, Faculty of Veterinary Medicine, Universiti Putra Malaysia, Serdang 43400, Malaysia; [email protected] 2 Department of Veterinary Pathology and Microbiology, Faculty of Veterinary Medicine, University of Nigeria Nsukka, Nsukka 410001, Nigeria 3 Animal Science Research Centre, Malaysian Agricultural Research and Development Institute, Headquarters, Serdang 43400, Malaysia; [email protected] 4 Department of Veterinary Pre-clinical sciences, Faculty of Veterinary Medicine, Universiti Putra Malaysia, Serdang 43400, Malaysia; [email protected] 5 Research Centre for Ruminant Diseases, Faculty of Veterinary Medicine, Universiti Putra Malaysia, Serdang 43400, Malaysia; [email protected] * Correspondence: [email protected] (O.A.A.); [email protected] (H.H.); Tel.: +60-11-352-01215 (O.A.A.); +60-19-284-6897 (H.H.) Received: 2 May 2020; Accepted: 16 July 2020; Published: 25 August 2020 Abstract: Theileriosis is a blood piroplasmic disease that adversely affects the livestock industry, especially in tropical and sub-tropical countries. It is caused by haemoprotozoan of the Theileria genus, transmitted by hard ticks and which possesses a complex life cycle. The clinical course of the disease ranges from benign to lethal, but subclinical infections can occur depending on the infecting Theileria species. The main clinical and clinicopathological manifestations of acute disease include fever, lymphadenopathy, anorexia and severe loss of condition, conjunctivitis, and pale mucous membranes that are associated with Theileria-induced immune-mediated haemolytic anaemia and/or non-regenerative anaemia. -
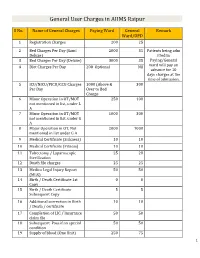
General User Charges in AIIMS Raipur
General User Charges in AIIMS Raipur S No. Name of General Charges Paying Ward General Remark Ward/OPD 1 Registration Charges 200 25 2 Bed Charges Per Day (Sami 2000 35 Patients being adm Deluxe) itted in 3 Bed Charges Per Day (Deluxe) 3000 35 Paying/General 4 Diet Charges Per Day 200 Optional Nil ward will pay an advance for 10 days charges at the time of admission. 5 ICU/NICU/PICU/CCU Charges 1000 (Above & 300 Per Day Over to Bed Charge 6 Minor Operation in OT/MOT 250 100 not mentioned in list, under L A 7 Minor Operation in OT/MOT 1000 300 not mentioned in list, under G A 8 Major Operation in OT, Not 2000 1000 mentioned in list under G A 9 Medical Certificate (Sickness) 10 10 10 Medical Certificate (Fitness) 10 10 11 Tubectomy / Laparoscopic 25 20 Sterilization 12 Death file charges 25 25 13 Medico Legal Injury Report 50 50 (MLR) 14 Birth / Death Certificate 1st 0 0 Copy 15 Birth / Death Certificate 5 5 Subsequent Copy 16 Additional correction in Birth 10 10 / Death / certificate 17 Completion of LIC / Insurance 50 50 claim file 18 Subsequent Pass if on special 50 50 condition 19 Supply of blood (One Unit) 250 75 1 20 Medical Board Certificate 500 500 On Special Case User Charges for Investigations in AIIMS Raipur S No. Name of Investigations Paying General Remark Ward Ward/OPD Anaesthsia 1 ABG 75 50 2 ABG ALONGWITH 150 100 ELECTROLYTES(NA+,K+)(Na,K) 3 ONLY ELECTROLYTES(Na+,K+,Cl,Ca+) 75 50 4 ONLY CALCIUM 50 25 5 GLUCOSE 25 20 6 LACTATE 25 20 7 UREA. -

Practice Parameter for the Diagnosis and Management of Primary Immunodeficiency
Practice parameter Practice parameter for the diagnosis and management of primary immunodeficiency Francisco A. Bonilla, MD, PhD, David A. Khan, MD, Zuhair K. Ballas, MD, Javier Chinen, MD, PhD, Michael M. Frank, MD, Joyce T. Hsu, MD, Michael Keller, MD, Lisa J. Kobrynski, MD, Hirsh D. Komarow, MD, Bruce Mazer, MD, Robert P. Nelson, Jr, MD, Jordan S. Orange, MD, PhD, John M. Routes, MD, William T. Shearer, MD, PhD, Ricardo U. Sorensen, MD, James W. Verbsky, MD, PhD, David I. Bernstein, MD, Joann Blessing-Moore, MD, David Lang, MD, Richard A. Nicklas, MD, John Oppenheimer, MD, Jay M. Portnoy, MD, Christopher R. Randolph, MD, Diane Schuller, MD, Sheldon L. Spector, MD, Stephen Tilles, MD, Dana Wallace, MD Chief Editor: Francisco A. Bonilla, MD, PhD Co-Editor: David A. Khan, MD Members of the Joint Task Force on Practice Parameters: David I. Bernstein, MD, Joann Blessing-Moore, MD, David Khan, MD, David Lang, MD, Richard A. Nicklas, MD, John Oppenheimer, MD, Jay M. Portnoy, MD, Christopher R. Randolph, MD, Diane Schuller, MD, Sheldon L. Spector, MD, Stephen Tilles, MD, Dana Wallace, MD Primary Immunodeficiency Workgroup: Chairman: Francisco A. Bonilla, MD, PhD Members: Zuhair K. Ballas, MD, Javier Chinen, MD, PhD, Michael M. Frank, MD, Joyce T. Hsu, MD, Michael Keller, MD, Lisa J. Kobrynski, MD, Hirsh D. Komarow, MD, Bruce Mazer, MD, Robert P. Nelson, Jr, MD, Jordan S. Orange, MD, PhD, John M. Routes, MD, William T. Shearer, MD, PhD, Ricardo U. Sorensen, MD, James W. Verbsky, MD, PhD GlaxoSmithKline, Merck, and Aerocrine; has received payment for lectures from Genentech/ These parameters were developed by the Joint Task Force on Practice Parameters, representing Novartis, GlaxoSmithKline, and Merck; and has received research support from Genentech/ the American Academy of Allergy, Asthma & Immunology; the American College of Novartis and Merck. -

Microbiology and Immunology
College of Medicine MI Microbiology and Immunology MI 494G IMMUNOBIOLOGY. (3) A survey of theories and mechanisms of immunity, including: nature of antigens and antibodies, antigen-antibody reactions, immunocompetent cells, immunogenetics, allergic reactions, tumor immunology and transplantation immunology. Prereq: BCH 401G (may be taken concurrently) and BIO 208 or BIO 308 or consent of instructor. (Same as BIO 494G.) MI 590 CELLULAR AND MOLECULAR PHYSIOLOGY. (4) This course will focus on the cellular and molecular physiology of inter-and intracellular communication. In particular, it will provide an overview of established and emerging intracellular signaling mechanisms which utilize i) cyclic nucleotides (cAMP; cGMP), ii) calcium (phosphatidylinositol metabolism: cyclic ADP-ribose), iii) transmembrane ion fluxes (voltage- and receptor-operated channels), iv) tyrosine kinases, and v) nuclear transcription factors. The material will be presented in a number of formats including didactic lecture and group discussions of selected readings. Prereq: PGY 412G, PGY 502 or consent of instructor. (Same as PGY 590.) MI 595 IMMUNOBIOLOGY LABORATORY. (2) Laboratory in immunology and serology. Preparation, standardization, and uses of biological products; serology. Laboratory; four hours. Prereq: BIO/MI 494G or concurrently; or consent of instructor. (Same as BIO 595.) MI 598 CLINICAL MICROBIOLOGY. (3) An introduction to the concepts of clinical microbiology through a survey of the microbial diseases of man using an organ system approach. Prereq: BIO 208 and 209, BIO 476G recommended, CHE 230 or 236, or consent of instructor. (Same as PAT 598.) MI 601 SPECIAL TOPICS IN MOLECULAR AND CELLULAR GENETICS. (1) Each semester five distinguished scientists visit the UK campus to deliver a series of three formal lectures each and participate in numerous informal contacts with graduate students. -

Level of Anti-FSH and Anti-LH Antibody in PCOS Women And
ry & Imm st u i n m o e p h a c t Hussein et al., Immunochem Immunopathol 2018, 4:1 o h o n Immunochemistry & l u o DOI: 10.4172/2469-9756.1000129 g m y m I ISSN: 2469-9756 Immunopathology ResearchResearch Article Article Open Access Level of Anti-FSH and Anti-LH Antibody in PCOS Women and Comparing it with Normal Control Group Shafaq Hussein*, Ihsan Al-Saimary and Maysoon Sherif Department of Microbiology, College of Medicine, University of Basrah, Iraq Abstract Background: Polycystic ovary syndrome is commonest endocrine disorder of female at reproductive age with estimation of its prevalence in general population was 20-33%. Objective: To estimate the levels of Anti-FSH Ab, Anti-LH Ab in polycystic women and comparing it with healthy control group. Patients and methods: This case controls study was carried out in Basrah. In this study blood sample from 29 women with PCOS and 29 healthy control women were collected from outpatient and private gynecological clinics and primary health care centers from different area of Basra from August 2016 to March 2018 for estimation of their serum levels of Anti-FSH Ab, Anti-LH Ab by using enzyme linked immunosorbent assay technique. Results: Serum levels of anti-FSH Ab, anti-LH Ab were elevated in PCOS women. Conclusion: Serum levels of anti-FSH Ab, anti-LH Ab is highly statistically significance in PCOS women than in healthy control group. Keywords: PCOS; Anti-FSH Ab; Anti-LH Ab; Serum levels comparing to normal control group. Introduction Materials and Methods Polycystic ovarian syndrome is a heterogeneous collection of Patients signs and symptoms that form a spectrum of a disorder with a mild A total number of 29 women with PCOS was involved in this case presentation in some women and a severe disturbance of reproductive, control study. -

Implications of Low-Grade Inflammation in SARS-Cov-2 Immunopathology
Review Article Implications of Low-grade Inflammation in SARS-CoV-2 Immunopathology Anamary Suárez-Reyes MD and Carlos A. Villegas-Valverde MD MS ABSTRACT senescent and obese patients and those with cardiovascular disease, INTRODUCTION Advanced age and chronic disease comorbidities cancer or chronic obstructive pulmonary disease. Low-grade chronic are indicators of poor prognosis in COVID-19 clinical progression. infl ammation is characteristic of all these conditions, refl ected in a pro- infl ammatory state, endothelial dysfunction, and changes to innate Fatal outcomes in patients with these characteristics are due to a immunity; mainly of the monocyte-macrophage system with changes dysfunctional immune response. Understanding COVID-19’s immu- in polarization, infl ammation, cytotoxicity and altered antigenic pre- nopathogenesis helps in designing strategies to prevent and mitigate sentation. In the case of SARS-CoV-2 infection, mechanisms involved complications during treatment. in acute infl ammation overlap with the patient’s pro-infl ammatory state, causing immune system dysfunction. SARS-CoV-2 infection OBJECTIVE Describe the main immunopathogenic alterations of amplifi es already-existing alterations, causing failures in the immune COVID-19 in patients of advanced age or with chronic non-commu- system’s control mechanisms. The resulting cytokine storm causes an nicable diseases. uncontrolled systemic infl ammatory response marked by high serum levels of infl ammatory biomarkers and a pro-infl ammatory cytokine DATA ACQUISITION We carried out a bibliographic search of pri- profi le with decompensation of underlying diseases. In asthma, chron- mary references in PubMed, Elsevier, Science Direct and SciELO. A ic eosinophilic infl ammation protects against infection by producing total of 270 articles met our initial search criteria. -

٢Year Dr. Hiyam Adil Altaii Lec. ٣ IMMUNOPATHOLOGY
2Year Dr. Hiyam Adil Altaii Lec. 3 IMMUNOPATHOLOGY IMMUNOPATHOLOGY The infected tissue often serves as an innocent bystander and immunopathology results. This can occur in acute and chronic infections. Over stimulation of cytokine production and complement activation by endotoxins can cause tissue injury in the absence of an immune response. Continuously generated antigens released from persisting viable microbes will subsequently elicit humoral antibodies and cell mediated immunity resulting in chronic immunopathology. Certain poorly degradable antigens (e.g pneumococcal polysaccharide and group A streptococcal cell walls) can maintain immunopathology even in the absence of persistence of live agents. Other bacterial antigens cross-react with host tissue antigens causing the development of autoimmunity (e.g. the M protein of S. pyogenes cross-reacts with mammalian myosin). Thus immunopathology can persist even after the infection and microbial antigens are eliminated. The immune system in resistance to infection - examples 1. Extracellular parasites. Antibodies cause lysis of the organism and/or their opsonization by phagocytes at which point they are rapidly killed. 2. Intracellular parasites are primarily killed by cell mediated immunity. 3. Exotoxins can be neutralized by antitoxins. These can be elicited using toxoid vaccines (toxoids are antigenic but not toxic). This occurs, for example, in vaccination against diphtheria. 4. Certain organisms produce IgA proteases (including H. influenzae, S. pneumoniae, N. gonorrhoeae and N. -

Medical Microbiology and Immunology 1
Medical Microbiology and Immunology 1 MEDICAL MICROBIOLOGY AND Doctor of Philosophy (Ph.D.) Program The objective of the program is to prepare highly qualified students for IMMUNOLOGY a broad range of possible careers in research and teaching in medical microbiology and immunology and related health science fields. Study Program Director: Patrick C. Swanson, Ph.D. for the Ph.D. degree emphasizes independence in scientific pursuit, Program Office: Criss II, Room 529 with a particular emphasis on research. Course work and dissertation research are designed to bring the student to a high-level of competence Graduate Study in Medical Microbiology in microbiology and immunology with particular expertise in the area chosen for dissertation research. You will be expected to demonstrate and Immunology a high capacity for original and independent thought, and apply this Within the context of Creighton as a Jesuit, Catholic University, the creativity, educational background, and knowledge of the scientific Graduate School provides value-centered education for students to method to dissertation research. develop mastery of their chosen field of study. The Medical Microbiology and Immunology programs offer an environment ideal for fostering Master of Science (M.S.) Program critical judgment, scholarly initiative, and disciplined inquiry. The objectives of the program include preparation of the student for one or more of the following careers: Program Goals At the completion of this graduate program in Medical Microbiology & 1. teaching of medical microbiology and immunology at the Immunology, students will: undergraduate level, and 2. participation in supervised or team research in universities, industry 1. Demonstrate advanced knowledge in the fields of Medical or government. -

Innate Versus Adaptive Immunity in Kidney Immunopathology Hans-Joachim Anders
Anders BMC Nephrology 2013, 14:138 http://www.biomedcentral.com/1471-2369/14/138 CORRESPONDENCE Open Access Innate versus adaptive immunity in kidney immunopathology Hans-Joachim Anders Abstract Most kidney disorders involve some degree of inflammation, i.e. induction of pro-inflammatory mediators and leukocyte recruitment. But what are the factors that determine inflammation as a trigger or a consequence of kidney injury? Which types of renal inflammation can be targeted by the novel more selective immunosuppressive and anti-inflammatory agents? How to dissect the mechanisms behind innate and adaptive immune responses that are orchestrated inside or outside the kidney but both cause renal immunopathology i.e. renal inflammation? How to dissect leukocytic cell infiltrates into pro-inflammatory leukocytes from anti-inflammatory and pro-regenerative leukocytes? How to dissect leukocytes that support epithelial repair from those that promote renal fibrosis. The term ‘renal inflammation’ has moved far beyond the descriptive category of ‘mixed leukocytic cell infiltrates’ as commonly described in kidney biopsies. It is time to face the complexity of renal inflammation to finally benefit from the new age of novel immunomodulatory medicines. Keywords: Glomerulonephritis, Acute kidney injury, Chronic kidney injury, Chemokines, Macrophages, B cells, T cells Inflammation is one of several danger response programs the implementation of organ transplantation into clinical that helps to control dangers that disrupt kidney homeo- practice [3]. stasis -

Immunopathology of Uveitis
British Journal of Ophthalmology 1998;82:91–96 91 PERSPECTIVE Br J Ophthalmol: first published as 10.1136/bjo.82.1.91 on 1 January 1998. Downloaded from Immunopathology of uveitis Chi-Chao Chan, Qian Li Using conventional histological (light and electron micro- surrounding the wound and foreign material. Tumours scopic examinations), immunohistological (immunofluo- may also initiate an inflammatory response. rescent and immunoperoxidase), and molecular histologi- Thirdly, the inflammatory process involves two types of cal (in situ hybridisation and polymerase chain reaction cellular components—the infiltrating inflammatory cells (PCR) in situ hybridisation) techniques, the immunopa- and the ocular resident cells. The types and subtypes of thology of uveitis has been studied using inflamed ocular inflammatory cells are easily identified by routine histology tissue.1–4 The findings usually provide helpful information and immunohistochemical stains.34 These cells release in the diagnosis and therapy of uveitis. The immunopa- numerous lymphokines, cytokines, immunoglobulins, thology of uveitis allows the visualisation of the morpho- growth factors, and inflammatory mediators, which can be logical interaction in the eye at the time the specimen is identified by immunohistochemistry.3 The messenger obtained. This information also helps in the understanding RNAs of many cytokines and growth factors can be of the immunopathogenesis of ocular inflammation. detected by molecular histological techniques.12The ocu- Three main aspects of pathological examinations are lar resident cells may undergo oedema, damage, necrosis, analysed. Firstly, the morphology of the ocular specimen or proliferation. They also respond by releasing cytokines, illustrates the lesions and specific localisations within the growth factors, and altering cellular markers including eye. -

Topics in Immunopathology 2021
TOPICS Immunopathology This document forms Appendix 7 to Trainee HandbookTOPICS for INImmunopathology IMMUNOPATHOLOGY and should be read in conjunction with Section 2 of the Handbook The document was prepared in April 2009 by A/Prof David Fulcher, with contributions from Dr Richard Wong, Dr Melanie Wong and A/Prof William Sewell and was revised in March 2011. RCPA - Topics in Immunopathology 1 Functions as a medical specialist in the laboratory 1.1. Knowledge and skills in immunobiology & pathogenesis of immunological disorders 1.1.1. Organisation of the immune system 1.1.1.1. Lymphoid tissues and organs: anatomy and function 1.1.1.2. Cells of relevance to the immune response, ontogeny, characteristics and functions 1.1.1.3. Molecules of relevance to the immune response: MHC, cytokines, chemokines, receptors, signalling pathways, vasoactive mediators, prostaglandins, leukotrienes 1.1.1.4. Genes of relevance to the immune response, including genes for above molecules and MHC, gene rearrangement in generation of antigen receptors 1.1.1.5. Structure and function relationships between the organs, tissues and cells that participate in immune responses 1.1.1.6. Cellular and molecular correlates of immune function 1.1.1.7. Comprehensive understanding of the ontogeny of the immune system and the relationship between the development of immune function to development of allergic diseases, autoimmunity, vasculitides and immunodeficiencies. 1.1.2. Immune mechanisms 1.1.2.1. Innate immunity: 1.1.2.1.1. NK cells 1.1.2.1.1.1. Cytokines relevant in NK cell development and function 1.1.2.1.1.2. -

Pathogenesis of Human Cytomegalovirus in the Immunocompromised Host
REVIEWS Pathogenesis of human cytomegalovirus in the immunocompromised host Paul Griffiths ✉ and Matthew Reeves Abstract | Human cytomegalovirus (HCMV) is a herpesvirus that infects ~60% of adults in developed countries and more than 90% in developing countries. Usually, it is controlled by a vigorous immune response so that infections are asymptomatic or symptoms are mild. However, if the immune system is compromised, HCMV can replicate to high levels and cause serious end organ disease. Substantial progress is being made in understanding the natural history and pathogenesis of HCMV infection and disease in the immunocompromised host. Serial measures of viral load defined the dynamics of HCMV replication and are now used routinely to allow intervention with antiviral drugs in individual patients. They are also used as pharmacodynamic read- outs to evaluate prototype vaccines that may protect against HCMV replication and to define immune correlates of this protection. This novel information is informing the design of randomized controlled trials of new antiviral drugs and vaccines currently under evaluation. In this Review, we discuss immune responses to HCMV and countermeasures deployed by the virus, the establishment of latency and reactivation from it, exogenous reinfection with additional strains, pathogenesis, development of end organ disease, indirect effects of infection, immune correlates of control of replication, current treatment strategies and the evaluation of novel vaccine candidates. Infectious mononucleosis Human cytomegalovirus (HCMV; also known as human overt symptoms, there is evidence that infected indi- A syndrome of fever and herpesvirus 5) is the prototype member of the Betaher- viduals may have long- term adverse outcomes related malaise with an excess of pesvirinae.