Acadian Variant of Fanconi Syndrome Is Caused by Mitochondrial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Large Meta-Analysis of Genome-Wide Association Studies
medRxiv preprint doi: https://doi.org/10.1101/2020.10.01.20200659; this version posted October 4, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Large meta-analysis of genome-wide association studies expands knowledge of the genetic etiology of Alzheimer’s disease and highlights potential translational opportunities Céline Bellenguez1,*,#, Fahri Küçükali2,3,4*, Iris Jansen5,6*, Victor Andrade7,8*, Sonia Morenau- Grau9,10,*, Najaf Amin11,12, Benjamin Grenier-Boley1, Anne Boland13, Luca Kleineidam7,8, Peter Holmans14, Pablo Garcia9,10, Rafael Campos Martin7, Adam Naj15,16, Yang Qiong17, Joshua C. Bis18, Vincent Damotte1, Sven Van der Lee5,6,19, Marcos Costa1, Julien Chapuis1, Vilmentas Giedraitis20, María Jesús Bullido10,21, Adolfo López de Munáin10,22, Jordi Pérez- Tur10,23, Pascual Sánchez-Juan10,24, Raquel Sánchez-Valle25, Victoria Álvarez26, Pau Pastor27, Miguel Medina10,28, Jasper Van Dongen2,3,4, Christine Van Broeckhoven2,3,4, Rik Vandenberghe29,30, Sebastiaan Engelborghs31,32, Gael Nicolas33, Florence Pasquier34, Olivier Hanon35, Carole Dufouil36, Claudine Berr37, Stéphanie Debette36, Jean-François Dartigues36, Gianfranco Spalletta38, Benedetta Nacmias39,40, Vincenzo Solfrezzi41, Barbara Borroni42, Lucio Tremolizzo43, Davide Seripa44, Paolo Caffarra45, Antonio Daniele46,47, Daniela Galimberti48,49, Innocenzo Rainero50, Luisa Benussi51, Alesio Squassina52, Patrizia Mecoci53, Lucilla Parnetti54, Carlo Masullo55, Beatrice Arosio56, John Hardy57, Simon Mead58, Kevin Morgan59, Clive Holmes60, Patrick Kehoe61, Bob Woods62, EADB, Charge, ADGC, Jin Sha15,16, Yi Zhao15,63, Chien-Yueh Lee15,63, Pavel P. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

A Large-Scale Multi-Ethnic Genome-Wide Association Study of Coronary Artery Disease
A large-scale multi-ethnic genome-wide association study of coronary artery disease Themistocles Assimes ( [email protected] ) Stanford University School of Medicine https://orcid.org/0000-0003-2349-0009 Catherine Tcheandjieu Stanford University Xiang Zhu The Pennsylvania State University https://orcid.org/0000-0003-1134-6413 Austin Hilliard VA Palo Alto Health Care System Shoa Clarke Stanford University School of Medicine https://orcid.org/0000-0002-6592-1172 Valerio Napolioni University of Camerino Shining Ma Stanford University School of Medicine Huaying Fang Stanford University School of Medicine Bryan R Gorman Massachusetts Veterans Epidemiology and Research Information Center, Veterans Affairs Boston Healthcare System Kyung Min Lee Edith Nourse Rogers Memorial Veterans Hospital https://orcid.org/0000-0001-8995-0448 Fei Chen University of Southern California Saiju Pyarajan Massachusetts Veterans Epidemiology and Research Information Center, Veterans Affairs Boston Healthcare System Rebecca Song VA Boston Healthcare System Mary Plomondon Rocky Mountain Regional VA Medical Center Thomas Maddox Washington University School of Medicine Stephen Waldo Rocky Mountain Regional VA Medical Center Nasa Sinnott-Armstrong Stanford University https://orcid.org/0000-0003-4490-0601 Yuk-Lam Ho Massachusetts Veterans Epidemiology and Research Information Center, Veterans Affairs Boston Healthcare System Genevieve Wojcik Johns Hopkins University Steven Buyske Rutgers University Charles Kooperberg Fred Hutchinson Cancer Research Center Jeffrey Haessler -

A Common Analgesic Enhances the Anti-Tumour Activity of 5-Aza-2’- Deoxycytidine Through Induction of Oxidative Stress
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.31.017947; this version posted April 1, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. A common analgesic enhances the anti-tumour activity of 5-aza-2’- deoxycytidine through induction of oxidative stress Hannah J. Gleneadie1,10, Amy H. Baker1, Nikolaos Batis2, Jennifer Bryant2, Yao Jiang3, Samuel J.H. Clokie4, Hisham Mehanna2, Paloma Garcia5, Deena M.A. Gendoo6, Sally Roberts5, Alfredo A. Molinolo7, J. Silvio Gutkind8, Ben A. Scheven1, Paul R. Cooper1, Farhat L. Khanim9 and Malgorzata Wiench1, 5,*. 1School of Dentistry, Institute of Clinical Studies, College of Medical and Dental Sciences, The University of Birmingham, Birmingham, B5 7EG, UK; 2Institute of Head and Neck Studies and Education (InHANSE), The University of Birmingham, Birmingham, B15 2TT, UK; 3School of Biosciences, The University of Birmingham, Birmingham, B15 2TT, UK; 4West Midlands Regional Genetics Laboratory, Birmingham Women’s and Children’s Hospital, Birmingham, B15 2TG, UK; 5Institute of Cancer and Genomic Sciences, College of Medical and Dental Sciences, The University of Birmingham, Birmingham, B15 2TT, UK; 6Centre for Computational Biology, Institute of Cancer and Genomic Sciences, The University of Birmingham, Birmingham, B15 2TT, UK; 7Moores Cancer Center and Department of Pathology, University of California San Diego, La Jolla, CA 92093, USA; 8Department of Pharmacology and Moores Cancer -

A Chromosome Level Genome of Astyanax Mexicanus Surface Fish for Comparing Population
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.06.189654; this version posted July 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title 2 A chromosome level genome of Astyanax mexicanus surface fish for comparing population- 3 specific genetic differences contributing to trait evolution. 4 5 Authors 6 Wesley C. Warren1, Tyler E. Boggs2, Richard Borowsky3, Brian M. Carlson4, Estephany 7 Ferrufino5, Joshua B. Gross2, LaDeana Hillier6, Zhilian Hu7, Alex C. Keene8, Alexander Kenzior9, 8 Johanna E. Kowalko5, Chad Tomlinson10, Milinn Kremitzki10, Madeleine E. Lemieux11, Tina 9 Graves-Lindsay10, Suzanne E. McGaugh12, Jeff T. Miller12, Mathilda Mommersteeg7, Rachel L. 10 Moran12, Robert Peuß9, Edward Rice1, Misty R. Riddle13, Itzel Sifuentes-Romero5, Bethany A. 11 Stanhope5,8, Clifford J. Tabin13, Sunishka Thakur5, Yamamoto Yoshiyuki14, Nicolas Rohner9,15 12 13 Authors for correspondence: Wesley C. Warren ([email protected]), Nicolas Rohner 14 ([email protected]) 15 16 Affiliation 17 1Department of Animal Sciences, Department of Surgery, Institute for Data Science and 18 Informatics, University of Missouri, Bond Life Sciences Center, Columbia, MO 19 2 Department of Biological Sciences, University of Cincinnati, Cincinnati, OH 20 3 Department of Biology, New York University, New York, NY 21 4 Department of Biology, The College of Wooster, Wooster, OH 22 5 Harriet L. Wilkes Honors College, Florida Atlantic University, Jupiter FL 23 6 Department of Genome Sciences, University of Washington, Seattle, WA 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.06.189654; this version posted July 6, 2020. -

Genome-Wide Profiling of Druggable Active Tumor Defense Mechanisms to Enhance Cancer Immunotherapy
bioRxiv preprint doi: https://doi.org/10.1101/843185; this version posted November 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Genome-wide profiling of druggable active tumor defense mechanisms to enhance cancer immunotherapy Rigel J. Kishton1,2,*,#, Shashank J. Patel1,2,†,*, Suman K. Vodnala1,2, Amy E. Decker3, Yogin Patel1,2, Madhusudhanan Sukumar1,2, Tori N. Yamamoto1,2,4, Zhiya Yu1,2, Michelle Ji1,2, Amanda N. Henning1,2, Devikala Gurusamy1,2, Douglas C. Palmer1,2, Winifred Lo1, Anna Pasetto1, Parisa Malekzadeh1, Drew C. Deniger1, Kris C. Wood3, Neville E. Sanjana5,6, Nicholas P. Restifo1,2, #, § 1Surgery Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD 20892, USA 2Center for Cell-Based Therapy, National Cancer Institute, Bethesda, MD 20892, USA 3Department of Pharmacology & Cancer Biology, Duke University School of Medicine, Durham, NC, USA 4Immunology Graduate Group, University of Pennsylvania, Philadelphia, PA 19104, USA 5New York Genome Center, New York, NY 10013 USA 6Department of Biology, New York University, New York, NY 10003, USA *These authors contributed equally to this work. †Present address: NextCure Inc., Beltsville, MD 20705, USA §Present address: Lyell Immunopharma, South San Francisco, CA 94080, USA #Corresponding authors. NPR: [email protected]. RJK: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/843185; this version posted November 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. -

NDUFAF6 (K-13): Sc-87001
SAN TA C RUZ BI OTEC HNOL OG Y, INC . NDUFAF6 (K-13): sc-87001 BACKGROUND PRODUCT Made up of nearly 146 million bases, chromosome 8 encodes about 800 genes. Each vial contains 100 µg IgG in 1.0 ml of PBS with < 0.1% sodium azide Translocation of portions of chromosome 8 with amplifications of the c-Myc and 0.1% gelatin. gene are found in some leukemias and lymphomas, and are typically associated Blocking peptide available for competition studies, sc-87001 P, (100 µg with a poor prognosis. Portions of chromosome 8 have been linked to schizo - peptide in 0.5 ml PBS containing < 0.1% sodium azide and 0.2% BSA). phrenia and bipolar disorder. Trisomy 8, also known as Warkany syndrome 2, most often results in early miscarriage but is occasionally seen in a mosaic APPLICATIONS form in surviving patients who suffer to a varying degree from a number of symptoms, including retarded mental and motor development, and certain NDUFAF6 (K-13) is recommended for detection of NDUFAF6 of human origin, facial and developmental defects. WRN is a DNA helicase encoded by chromo - 2310030N02Rik of mouse origin and the corresponding rat homolog by some 8 and shown defective in those with the early aging disorder Werner Western Blotting (starting dilution 1:200, dilution range 1:100-1:1000), syndrome. Chromosome 8 is also associated with Pfeiffer syndrome, congeni - immunoprecipitation [1-2 µg per 100-500 µg of total protein (1 ml of cell tal hypothyroidism and Waardenburg syndrome. The C8orf38 gene product has lysate)], immunofluorescence (starting dilution 1:50, dilution range 1:50- been implicated as a cause of mitochondrial complex I deficiency (MT-C1D), 1:500) and solid phase ELISA (starting dilution 1:30, dilution range 1:30- disorder of the mitochondrial respiratory chain that causes a wide range of 1:3000). -

Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions
International Journal of Molecular Sciences Review Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions Margherita Protasoni 1 and Massimo Zeviani 1,2,* 1 Mitochondrial Biology Unit, The MRC and University of Cambridge, Cambridge CB2 0XY, UK; [email protected] 2 Department of Neurosciences, University of Padova, 35128 Padova, Italy * Correspondence: [email protected] Abstract: Mitochondria are ubiquitous intracellular organelles found in almost all eukaryotes and involved in various aspects of cellular life, with a primary role in energy production. The interest in this organelle has grown stronger with the discovery of their link to various pathologies, including cancer, aging and neurodegenerative diseases. Indeed, dysfunctional mitochondria cannot provide the required energy to tissues with a high-energy demand, such as heart, brain and muscles, leading to a large spectrum of clinical phenotypes. Mitochondrial defects are at the origin of a group of clinically heterogeneous pathologies, called mitochondrial diseases, with an incidence of 1 in 5000 live births. Primary mitochondrial diseases are associated with genetic mutations both in nuclear and mitochondrial DNA (mtDNA), affecting genes involved in every aspect of the organelle function. As a consequence, it is difficult to find a common cause for mitochondrial diseases and, subsequently, to offer a precise clinical definition of the pathology. Moreover, the complexity of this condition makes it challenging to identify possible therapies or drug targets. Keywords: ATP production; biogenesis of the respiratory chain; mitochondrial disease; mi-tochondrial electrochemical gradient; mitochondrial potential; mitochondrial proton pumping; mitochondrial respiratory chain; oxidative phosphorylation; respiratory complex; respiratory supercomplex Citation: Protasoni, M.; Zeviani, M. -

Genetics of Abca4-Associated Diseases and Retinitis Pigmentosa
GENETICS OF ABCA4-ASSOCIATED DISEASES AND RETINITIS PIGMENTOSA Yajing (Angela) Xie Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 © 2016 Yajing (Angela) Xie All rights reserved ABSTRACT Genetics of ABCA4-Associated Diseases and Retinitis Pigmentosa Yajing (Angela) Xie Inherited retinal dystrophies encompass a broad group of genetic disorders affecting visual functions in as high as 1 in 3,000 individuals around the world. Common symptoms include loss of central, periphery, or night visions, and in severe cases progression to complete blindness. Syndromic forms also exist involving abnormalities in other parts of the body. Currently, more than 250 genes representing a wide variety of functional roles have been shown to be responsible for the disease phenotypes. Moreover, mutations in the same gene sometimes cause different phenotypes while mutations in multiple genes can give rise to the same clinical subtype, further demonstrating the level of complexity in these disorders. Such genetic heterogeneity has substantially complicated the process of pinpointing precise genetic causes underlying these conditions. The goal of my thesis research is to clarify the genetic causes underlying retinal dystrophies, with a primary focus on phenotypes resembling ABCA4-associated diseases and retinitis pigmentosa in both syndromic and non-syndromic forms. Recent advances in the next-generation sequencing (NGS), the high-throughput, ‘deep’ sequencing technology, have enabled several novel genes to be identified, or found new mutations in known genes. Nevertheless, a substantial fraction of unsolved cases still remain. The primary work in this thesis involves utilizing NGS, particularly whole- exome sequencing, to identify disease-causal mutations in families where at least one parent and affected or unaffected siblings are available. -
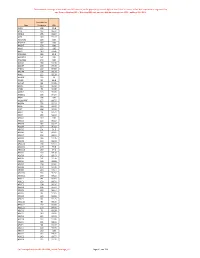
Onderstaande Coverage Is Berekend Over 860 Exomen, Welke Geprept Zijn Met De Agilent Sureselect XT Exome V7 Kit
Onderstaande coverage is berekend over 860 exomen, welke geprept zijn met de Agilent SureSelect XT exome v7 kit. Het sequencen is uitgevoerd op een Illumina NextSeq500 of NovaSeq6000 met een gemiddelde coverage van 100X , dekking 20x >93%. Gemiddelde Gen Coverage 20x A1BG 128 99.8 A1CF 132 99.32 A2ML1 155 99.97 A2M 148 99.72 A3GALT2 119 100 A4GALT 157 100 A4GNT 178 100 AAAS 123 100 AACS 143 97.96 AADACL2 186 99.9 AADACL3 161 100 AADACL4 170 100 AADAC 160 99.98 AADAT 138 97.39 AAED1 110 84.89 AAGAB 164 99.29 AAK1 122 99.39 AAMDC 135 90 AAMP 95 98.4 AANAT 86 99.85 AAR2 120 98.41 AARD 75 99.06 AARS2 123 99.89 AARSD1 115 94.24 AARS 134 100 AASDHPPT 157 99.75 AASDH 135 99.29 AASS 143 99.67 AATF 156 99.95 AATK 79 92.71 ABAT 136 99.16 ABCA1 149 100 ABCA2 123 96.52 ABCA3 132 99.27 ABCA4 137 96.03 ABCA5 101 94.3 ABCA6 131 98.41 ABCA7 102 99.12 ABCA8 146 99.26 ABCA9 140 99.34 ABCA10 126 94.11 ABCA12 159 99.8 ABCA13 159 97.2 ABCB1 160 99.58 ABCB4 142 99.43 ABCB5 151 99.76 ABCB6 149 99.98 ABCB7 128 99.81 ABCB8 100 93.36 ABCB9 124 97.14 ABCB10 136 90.97 ABCB11 158 99.22 ABCC1 141 96.65 ABCC2 150 99.71 ABCC3 108 96.16 ABCC4 134 95.97 ABCC5 130 96.14 ABCC6 96 92.85 ABCC8 136 99.83 ABCC9 141 99.75 ABCC10 90 99.35 ABCC11 117 99.93 ABCC12 153 99.93 ABCD1 74 83.58 ABCD2 143 99.92 ABCD3 122 92.63 ABCD4 122 99.84 ABCE1 141 98.18 ABCF1 90 99.42 ABCF2 128 99.93 ABCF3 159 100 ABCG1 135 99.71 ABCG2 143 99.77 ABCG4 122 99.91 De Coverage komt vanuit LAB-F0680_Exoom Coverage_v3. -

A Primer Genetic Toolkit for Exploring Mitochondrial Biology and Disease Using
bioRxiv preprint doi: https://doi.org/10.1101/542084; this version posted February 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 A primer genetic toolkit for exploring mitochondrial biology and disease using 2 zebrafish 3 Ankit Sabharwal1, Jarryd M. Campbell1,2, Zachary WareJoncas1, Mark Wishman1, Hirotaka 4 Ata1,2, Wiebin Liu1.3, Noriko Ichino1, Jake D. Bergren1, Mark D. Urban1, Rhianna Urban1, Tanya 5 L. Poshusta1, Yonghe Ding1,3, Xiaolei Xu1,3, Karl J. Clark1,2, Stephen C. Ekker1,2* 6 1. Department of Biochemistry and Molecular Biology, Mayo Clinic, Rochester, MN 55905, 7 USA 8 2. Center for Clinical and Translational Sciences, Mayo Clinic, Rochester, MN 55905, USA 9 3. Division of Cardiovascular Diseases, Mayo Clinic, Rochester, MN 55905, USA 10 *Corresponding author: [email protected] 11 Keywords: 12 Mitochondria, Mitochondrial disorders, Zebrafish, gene editing, 13 TALEN, Gene Breaking Transposon 1 bioRxiv preprint doi: https://doi.org/10.1101/542084; this version posted February 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 14 Abstract 15 Mitochondria are a dynamic eukaryotic innovation that play diverse roles in biology and disease. 16 The mitochondrial genome is remarkably conserved in all vertebrates, encoding the same 37 17 gene set and overall genomic structure ranging from 16,596 base pairs (bp) in the teleost 18 zebrafish (Danio rerio) to 16,569 bp in humans.