Recent Advances in Hepatitis B Treatment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
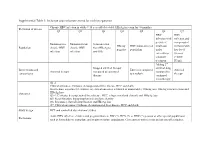
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

Tanibirumab (CUI C3490677) Add to Cart
5/17/2018 NCI Metathesaurus Contains Exact Match Begins With Name Code Property Relationship Source ALL Advanced Search NCIm Version: 201706 Version 2.8 (using LexEVS 6.5) Home | NCIt Hierarchy | Sources | Help Suggest changes to this concept Tanibirumab (CUI C3490677) Add to Cart Table of Contents Terms & Properties Synonym Details Relationships By Source Terms & Properties Concept Unique Identifier (CUI): C3490677 NCI Thesaurus Code: C102877 (see NCI Thesaurus info) Semantic Type: Immunologic Factor Semantic Type: Amino Acid, Peptide, or Protein Semantic Type: Pharmacologic Substance NCIt Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor tyrosine kinase expressed by endothelial cells, while VEGF is overexpressed in many tumors and is correlated to tumor progression. PDQ Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor -

Clevudine Induced Mitochondrial Myopathy
CROSSMARK_logo_3_Test 1 / 1 ORIGINAL ARTICLE Neuroscience https://crossmarhttps://doi.org/10.3346/jkms.2017.32.11.1857k-cdn.crossref.org/widget/v2.0/logos/CROSSMARK_Color_square.svg 2017-03-16 • J Korean Med Sci 2017; 32: 1857-1860 Clevudine Induced Mitochondrial Myopathy Soo-Hyun Park,1 Kyung-Seok Park,2 Clevudine was approved as an antiviral agent for hepatitis B virus, which showed marked, Nam-Hee Kim,1 Joong-Yang Cho,3 rapid inhibition of virus replication without significant toxicity. However, several studies Moon Soo Koh,4 and Jin Ho Lee4 have reported myopathy associated with clevudine therapy. Also, we experienced seven patients who suffered from myopathy during clevudine therapy. To characterize clevudine- 1Department of Neurology, Dongguk University Ilsan Hospital, Goyang, Korea; 2Department of Neurology, induced myopathy, we collected previously reported cases of clevudine myopathy and Seoul National University College of Medicine, analyzed all the cases including our cases. We searched electronic databases that were Seoul, Korea; 3Department of Neurology, Inje published in English or Korean using PubMed and KoreaMed. Ninety-five cases with University Ilsan Paik Hospital, Inje University College clevudine myopathy, including our seven cases, were selected and analyzed for the of Medicine, Goyang, Korea; 4Department of Hepatology, Dongguk University Ilsan Hospital, demographic data, clinical features, and pathologic findings. The 95 patients with Goyang, Korea clevudine-induced myopathy comprised 52 women and 43 men aged 48.9 years (27–76 years). The patients received clevudine therapy for about 14.2 months (5–24 months) Received: 20 January 2017 before the development of symptoms. Weakness mainly involved proximal extremities, Accepted: 29 July 2017 especially in the lower extremities, and bulbar and neck weakness were observed in some Address for Correspondence: cases (13.7%). -

DNA/RNA Synthesis
DNA/RNA Synthesis RNA synthesis, which is also called DNA transcription, is a highly selective process. Transcription by RNA polymerase II extends beyond RNA synthesis, towards a more active role in mRNA maturation, surveillance and export to the cytoplasm. Single-strand breaks are repaired by DNA ligase using the complementary strand of the double helix as a template, with DNA ligase creating the final phosphodiester bond to fully repair the DNA.DNA ligases discriminate against substrates containing RNA strands or mismatched base pairs at positions near the ends of the nickedDNA. Bleomycin (BLM) exerts its genotoxicity by generating free radicals, whichattack C-4′ in the deoxyribose backbone of DNA, leading to opening of the ribose ring and strand breakage; it is an S-independentradiomimetic agent that causes double-strand breaks in DNA. First strand cDNA is synthesized using random hexamer primers and M-MuLV Reverse Transcriptase (RNase H). Second strand cDNA synthesis is subsequently performed using DNA Polymerase I and RNase H. The remaining overhangs are converted into blunt ends using exonuclease/polymerase activity. After adenylation of the 3′ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure is ligated to prepare the samples for hybridization. Cell cycle and DNA replication are the top two pathways regulated by BET bromodomain inhibition. Cycloheximide blocks the translation of mRNA to protein. www.MedChemExpress.com 1 DNA/RNA Synthesis Inhibitors, Agonists, Activators, Modulators & Chemicals (+)-TK216 (-)-TK216 Cat. No.: HY-122903B Cat. No.: HY-122903A (+)-TK216 is an enantiomer of TK216 (HY-122903). (-)-TK216 is an enantiomer of TK216 (HY-122903). TK216 is an orally active and potent E26 TK216 is an orally active and potent E26 transformation specific (ETS) inhibitor. -
![Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set](https://docslib.b-cdn.net/cover/8870/ehealth-dsi-ehdsi-v2-2-2-or-ehealth-dsi-master-value-set-1028870.webp)
Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set
MTC eHealth DSI [eHDSI v2.2.2-OR] eHealth DSI – Master Value Set Catalogue Responsible : eHDSI Solution Provider PublishDate : Wed Nov 08 16:16:10 CET 2017 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 1 of 490 MTC Table of Contents epSOSActiveIngredient 4 epSOSAdministrativeGender 148 epSOSAdverseEventType 149 epSOSAllergenNoDrugs 150 epSOSBloodGroup 155 epSOSBloodPressure 156 epSOSCodeNoMedication 157 epSOSCodeProb 158 epSOSConfidentiality 159 epSOSCountry 160 epSOSDisplayLabel 167 epSOSDocumentCode 170 epSOSDoseForm 171 epSOSHealthcareProfessionalRoles 184 epSOSIllnessesandDisorders 186 epSOSLanguage 448 epSOSMedicalDevices 458 epSOSNullFavor 461 epSOSPackage 462 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 2 of 490 MTC epSOSPersonalRelationship 464 epSOSPregnancyInformation 466 epSOSProcedures 467 epSOSReactionAllergy 470 epSOSResolutionOutcome 472 epSOSRoleClass 473 epSOSRouteofAdministration 474 epSOSSections 477 epSOSSeverity 478 epSOSSocialHistory 479 epSOSStatusCode 480 epSOSSubstitutionCode 481 epSOSTelecomAddress 482 epSOSTimingEvent 483 epSOSUnits 484 epSOSUnknownInformation 487 epSOSVaccine 488 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 3 of 490 MTC epSOSActiveIngredient epSOSActiveIngredient Value Set ID 1.3.6.1.4.1.12559.11.10.1.3.1.42.24 TRANSLATIONS Code System ID Code System Version Concept Code Description (FSN) 2.16.840.1.113883.6.73 2017-01 A ALIMENTARY TRACT AND METABOLISM 2.16.840.1.113883.6.73 2017-01 -

Hepatitis B and Hepatitis D Luis S
Hepatitis B and Hepatitis D Luis S. Marsano, MD Professor of Medicine Division of Gastroenterology, Hepatology and Nutrition University of Louisville and Louisville VAMC June 2020 Hepatitis B Hepatitis B • 42 nm, partially double-stranded circular DNA virus. • 250 million carriers world-wide; – causes 500000 to 1 million deaths a year (686,000 in 2013) • 1.25 million carriers in USA.(0.5 %); – > 8% in Alaskan Eskimos. • Represents 5-10% of liver transplants worldwide. • New infections: decreasing in frequency – 260,000/y in 1980’s; – now 73,000/y • Greatest decline among children & adolescents (vaccine effect). Hepatitis B • Highest rate of disease in 20 to 49 year-olds • 20-30% of chronically infected americans acquired infection in childhood. • High prevalence in: – Asian-Pacific with 5-15% HBsAg(+) – Eastern European immigrants • Transmission: – In USA predominantly sexual and percutaneous during adult age. – In Alaska predominantly perinatal. Epidemiology and public health burden1 • Worldwide ≈250 million chronic HBsAg carriers2,3 • 686,000 deaths from HBV-related liver disease and HCC in 20134 HBsAg prevalence, adults (19−49 years), 20053 <2% Decreasing prevalence 2−4% in some endemic countries, e.g. Taiwan7 5−7% Possible reasons: ≥8% • Improved Not applicable socioeconomic status • Vaccination • Effective treatments Increasing prevalence in some European countries:5,6 • Migration from high endemic countries 1. EASL CPG HBV. J Hepatol 2017;67:370–98; 2. Schweitzer A, et al. Lancet 2015;386:1546–55; 3. Ott JJ, et al. Vaccine 2012;30:2212–9; 4. GBD 2013 Mortality and Causes of Death Collaborators. Lancet 2015;385:117–71; 5. Coppola N, et al. -

Appendices: V Ervolgonderzoek Medicatieveiligheid
APPENDICES: V ERVOLGONDERZOEK MEDICATIEVEILIGHEID Dit is een bijlage bij het rapport Vervolgonderzoek Medicatieveiligheid en is opgesteld voor het Ministerie van VWS vanuit een samenwerkingsverband tussen het Erasmus MC (Rotterdam), NIVEL (Utrecht), Radboud UMC (Nijmegen) en PHARMO (Utrecht) Januari 2017 Versie 1.0 1 Appendices Hoofstuk 2: Onderzoek naar de mate van opvolging van HARM-Wrestling aanbevelingen (2009-2014) 2 Appendix 1 Appendix 1: Technische omzetting van HARM-Wrestling aanbevelingen naar indicatoren Algemene specificaties Tabel A1a. Bepaling van medicatiegebruik. Geneesmiddel of ATC-code geneesmiddelen groep Antidepressiva N06A Laag gedoseerd ASA B01AC06, B01AC08, B01AC30, N02BA15 (dosering 100mg) of N02BA01 (dosering 80mg) Benzodiazepinen N05CF, N05CD, N05BA of N05CC Beta-blokkers C07 Bisfosfonaten M05BA, M05BB, of M05XX Calcineurine remmers L04AA05 of L04AD01 Carbamazepine N03AF01 Corticosteroiden H02AB Co-trimoxazol J01EE01 Coxibs M01AH Diabetesmedicatie A10 Digoxine C01AA05 Glibenclamide A10BB01 of A10BD02 of A10BD04 H2RA A02BA Itraconazol J02AC02 Kaliumsparende diuretica C03DA, C03DB, of C03EA Kaliumverliezende diuretica C03A, C03B, C03E, C07B, C07CB03, C09BA, C09DA, C09XA52, C03C of C09DX01 Ketoconazol J02AB02 Niet-selectieve NSAID’s N02BA01, N02BA15, N02BA11, N02BA51, N02BA65 of M01A met uitzondering van M01AH, M01AX05, M01AX12, M01AX21, M01AX24, M01AX25 en M01AX26 Laxantia A06A, A02AA02, A02AA03, A02AA04, A06AC, A06AA, of A06AG Lisdiuretica C03C Macroliden J01FA of A02BD04 VKA B01AA Opioïden N02AA met uitzondering van N02AA55, N02AA59 en N02AA79, N02AB, N02AC, N02AD, N02AG, N02AE of N07BC01 Pentamidine P01CX01 PPI’s A02BC of M01AE52 RAS-remmers C09 Sotalol C07AA07 Spironolacton C03DA01 SSRI's N06AB, N06AX21 of N06AX16 Sulonylureumderivaten A10BB, A10BD02 of A10BD04 Thiazidediuretica C03A, C03B, C03EA, C07B, C09BA, C09DA, C09XA52, C09DX01 of C07CB03 TAR B01AC04, B01AC06, B01AC08, B01AC22, B01AC30, N02BA15 (dosering 100mg) of N02BA01 (dosering 80mg) Thienopyridine derivaten B01AC04, B01AC22 of B01AC30 3 Appendix 1 Tabel A1b. -

New Approaches to the Treatment of Chronic Hepatitis B
Journal of Clinical Medicine Review New Approaches to the Treatment of Chronic Hepatitis B Alexandra Alexopoulou 1,*, Larisa Vasilieva 1 and Peter Karayiannis 2 1 Department of Medicine, Medical School, National & Kapodistrian University of Athens, Hippokration General Hospital, 11527 Athens, Greece; [email protected] 2 Department of Basic and Clinical Sciences, Medical School, University of Nicosia, Engomi, CY-1700 Nicosia, Cyprus; [email protected] * Correspondence: [email protected]; Tel.: +30-2132-088-178; Fax: +30-2107-706-871 Received: 3 September 2020; Accepted: 28 September 2020; Published: 1 October 2020 Abstract: The currently recommended treatment for chronic hepatitis B virus (HBV) infection achieves only viral suppression whilst on therapy, but rarely hepatitis B surface antigen (HBsAg) loss. The ultimate therapeutic endpoint is the combination of HBsAg loss, inhibition of new hepatocyte infection, elimination of the covalently closed circular DNA (cccDNA) pool, and restoration of immune function in order to achieve virus control. This review concentrates on new antiviral drugs that target different stages of the HBV life cycle (direct acting antivirals) and others that enhance both innate and adaptive immunity against HBV (immunotherapy). Drugs that block HBV hepatocyte entry, compounds that silence or deplete the cccDNA pool, others that affect core assembly, agents that degrade RNase-H, interfering RNA molecules, and nucleic acid polymers are likely interventions in the viral life cycle. In the immunotherapy category, molecules that activate the innate immune response such as Toll-like-receptors, Retinoic acid Inducible Gene-1 (RIG-1) and stimulator of interferon genes (STING) agonists or checkpoint inhibitors, and modulation of the adaptive immunity by therapeutic vaccines, vector-based vaccines, or adoptive transfer of genetically-engineered T cells aim towards the restoration of T cell function. -

Surveillance of Antimicrobial Consumption in Europe 2013-2014 SURVEILLANCE REPORT
SURVEILLANCE REPORT SURVEILLANCE REPORT Surveillance of antimicrobial consumption in Europe in Europe consumption of antimicrobial Surveillance Surveillance of antimicrobial consumption in Europe 2013-2014 2012 www.ecdc.europa.eu ECDC SURVEILLANCE REPORT Surveillance of antimicrobial consumption in Europe 2013–2014 This report of the European Centre for Disease Prevention and Control (ECDC) was coordinated by Klaus Weist. Contributing authors Klaus Weist, Arno Muller, Ana Hoxha, Vera Vlahović-Palčevski, Christelle Elias, Dominique Monnet and Ole Heuer. Data analysis: Klaus Weist, Arno Muller and Ana Hoxha. Acknowledgements The authors would like to thank the ESAC-Net Disease Network Coordination Committee members (Marcel Bruch, Philippe Cavalié, Herman Goossens, Jenny Hellman, Susan Hopkins, Stephanie Natsch, Anna Olczak-Pienkowska, Ajay Oza, Arjana Tambić Andrasevic, Peter Zarb) and observers (Jane Robertson, Arno Muller, Mike Sharland, Theo Verheij) for providing valuable comments and scientific advice during the production of the report. All ESAC-Net participants and National Coordinators are acknowledged for providing data and valuable comments on this report. The authors also acknowledge Gaetan Guyodo, Catalin Albu and Anna Renau-Rosell for managing the data and providing technical support to the participating countries. Suggested citation: European Centre for Disease Prevention and Control. Surveillance of antimicrobial consumption in Europe, 2013‒2014. Stockholm: ECDC; 2018. Stockholm, May 2018 ISBN 978-92-9498-187-5 ISSN 2315-0955 -

(12) United States Patent (10) Patent No.: US 8,158,152 B2 Palepu (45) Date of Patent: Apr
US008158152B2 (12) United States Patent (10) Patent No.: US 8,158,152 B2 Palepu (45) Date of Patent: Apr. 17, 2012 (54) LYOPHILIZATION PROCESS AND 6,884,422 B1 4/2005 Liu et al. PRODUCTS OBTANED THEREBY 6,900, 184 B2 5/2005 Cohen et al. 2002fOO 10357 A1 1/2002 Stogniew etal. 2002/009 1270 A1 7, 2002 Wu et al. (75) Inventor: Nageswara R. Palepu. Mill Creek, WA 2002/0143038 A1 10/2002 Bandyopadhyay et al. (US) 2002fO155097 A1 10, 2002 Te 2003, OO68416 A1 4/2003 Burgess et al. 2003/0077321 A1 4/2003 Kiel et al. (73) Assignee: SciDose LLC, Amherst, MA (US) 2003, OO82236 A1 5/2003 Mathiowitz et al. 2003/0096378 A1 5/2003 Qiu et al. (*) Notice: Subject to any disclaimer, the term of this 2003/OO96797 A1 5/2003 Stogniew et al. patent is extended or adjusted under 35 2003.01.1331.6 A1 6/2003 Kaisheva et al. U.S.C. 154(b) by 1560 days. 2003. O191157 A1 10, 2003 Doen 2003/0202978 A1 10, 2003 Maa et al. 2003/0211042 A1 11/2003 Evans (21) Appl. No.: 11/282,507 2003/0229027 A1 12/2003 Eissens et al. 2004.0005351 A1 1/2004 Kwon (22) Filed: Nov. 18, 2005 2004/0042971 A1 3/2004 Truong-Le et al. 2004/0042972 A1 3/2004 Truong-Le et al. (65) Prior Publication Data 2004.0043042 A1 3/2004 Johnson et al. 2004/OO57927 A1 3/2004 Warne et al. US 2007/O116729 A1 May 24, 2007 2004, OO63792 A1 4/2004 Khera et al. -

Malaysian Statistics on Medicines 2007
MALAYSIAN STATISTICS ON MEDICINES 2007 Edited by: Faridah AMY, Sivasampu S, Lian LM, Hazimah H, Kok LC, Chinniah RJ With contributions from Lian LM, Tang RY, Hafizh AA, Hazimah H, Gan HH, Kok LC, Leow AY, Lim JY, Thoo S, Hoo LP, Faridah AMY, Lim TO, Sivasampu S, Nour Hanah O, Fatimah AR, Nadia Fareeda MG, Goh A, Rosaida MS, Menon J, Radzi H, Yung CL, Michelle Tan HP, Yip KF, Chinniah RJ, Khutrun Nada Z, Masni M, Sri Wahyu T, Jalaludin MY, Leow NCW, Norafidah I, G R Letchuman R, Fuziah MZ, Mastura I, Sukumar R, Yong SL, Lim YX, Yap PK, Lim YS, Sujatha S, Goh AS, Chang KM, Wong SP, Omar I, Alan Fong YY, David Quek KL, Feisul IM, Long MS, Christopher Ong WM, Ghazali Ahmad, Abdul Rashid Abdul Rahman, Hooi LS, Khoo EM, Sunita Bavanandan, Nur Salima Shamsudin, Puteri Juanita Zamri, Sim KH, Wan Azman WA, Abdul Kahar AG, Faridah Y, Sahimi M, Haarathi C, Nirmala J, Azura MA, Asmah J, Rohna R, Choon SE, Roshidah B, Hasnah Z, Wong CW, Noorsidah MY, Sim HY, J Ravindran, Nik Nasri, Ghazali I, Wan Abu Bakar, Tham SW, J Ravichandran, Zaridah S, W Zahanim WY, Intan SS, Tan AL, Malek R, Sothilingam S, Syarihan S, Foo LK, Low KS, Janet YH Hong, Tan ATB, Lim PC, Loh YF, Nor Azizah, Sim BLH, Mohd Daud CY, Sameerah SAR, Muhd Nazri, Cheng JT, Lai J, Rahela AK, Lim GCC, Azura D, Rosminah MD, Kamarun MK, Nor Saleha IT, Tajunisah ME, Wong HS, Rosnawati Yahya, Manjulaa DS, Norrehan Abdullah, H Hussein, H Hussain, Salbiah MS, Muhaini O, Low YL, Beh PK, Cardosa MS, Choy YC, Lim RBL, Lee AW, Choo YM, Sapiah S, Fatimah SA, Norsima NS, Jenny TCN, Hanip R, Siti Nor -

Gilead to Acquire MYR Pharmaceuticals
Gilead to Acquire MYR Pharmaceuticals December 9, 2020 Forward-Looking Statements This presentation includes forward-looking statements, within the meaning of the Private Securities Litigation Reform Act of 1995, related to Gilead, MYR Pharmaceuticals and the acquisition of MYR Pharmaceuticals by Gilead that are subject to risks, uncertainties and other factors, including Gilead’s ability to successfully execute its corporate strategy in its currently anticipated timelines; Gilead’s ability to make progress on any of its long-term ambitions laid out in its corporate strategy; Gilead’s ability to accelerate or sustain revenues for its programs; the ability of the parties to complete the transaction in a timely manner or at all; the possibility that various closing conditions for the transaction may not be satisfied or waived, including the possibility that a governmental entity may prohibit, delay or refuse to grant approval for the consummation of the transaction; uncertainties relating to the timing or outcome of any filings and approvals relating to the transaction; difficulties or unanticipated expenses in connection with integrating the companies, including the effects of the transaction on relationships with employees, other business partners or governmental entities; the risk that Gilead may not realize the expected benefits of this transaction; the ability of Gilead to advance MYR Pharmaceuticals’ product pipeline and successfully commercialize Hepcludex®; the ability of the parties to initiate and complete clinical trials involving Hepcludex in the currently anticipated timelines or at all; the possibility of unfavorable results from one or more of such trials involving Hepcludex; uncertainties relating to regulatory applications and related filing and approval timelines, including the risk that the U.S.