Dissertation Tony E
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Novichok Agent - Wikipedia
18-3-2018 Novichok agent - Wikipedia Novichok agent Novichok (Russian: Новичо́к, "newcomer") is a series of nerve agents the Soviet Union and Russia developed between 1971 and 1993.[a][2][3] Russian scientists who developed the agents claim they are the deadliest nerve agents ever made, with some variants possibly five to eight times more potent than VX,[4][5] and others up to ten times more potent than soman.[6] They were designed as part of a Soviet program codenamed "FOLIANT".[7][1] Five Novichok variants are believed to have been weaponised for military use.[8] The most versatile was A-232 (Novichok-5).[9] Novichok agents have never been used on the battlefield. Theresa May, Prime Minister of the United Kingdom, said that one such agent was used in the poisoning of Sergei and Yulia Skripal in England in March 2018.[10] Russia officially denies producing or researching Novichok agents.[11] In 2013, the Organisation for the Prohibition of Chemical Weapons Scientific Advisory Board reported that it had insufficient information to comment on the existence or properties of Novichok agents,[12] and in 2011 it noted there was no peer reviewed paper on Novichok agents in scientific literature.[13] Contents Design objectives Disclosure Development and test sites Description of Novichok agents Chemistry Effects Use Poisoning of Kivelidi Poisoning of Sergei and Yulia Skripal See also References Further reading External links Design objectives These agents were designed to achieve four objectives:[14][15] To be undetectable using standard 1970s and 1980s NATO chemical detection equipment; To defeat NATO chemical protective gear; To be safer to handle; To circumvent the Chemical Weapons Convention list of controlled precursors, classes of chemical and physical form. -

Verification of Chemical Warfare Agent Exposure in Human Samples
Toxichem Krimtech 2013;80(Special Issue):288 Verification of chemical warfare agent exposure in human samples Paul W. Elsinghorst, Horst Thiermann, Marianne Koller Institut für Pharmakologie und Toxikologie der Bundeswehr, München Abstract Aim: This brief presentation provides an overview of methods that have been developed for the verification of human exposure to chemical warfare agents. Methods: GC–MS detection of nerve agents (V- and G-type) has been carried out with respect to unreacted agents as well as enzyme-bound species and metabolites. Methods involving di- rect SPE from plasma, fluoride-induced release of protein-bound nerve agents in plasma and analysis of their metabolites in plasma and urine have been developed. Exposure to blistering agents, i.e., sulfur mustard, has been verified by GC–MS detection of the unreacted agent in plasma and by LC– and GC–MS analysis of its metabolites in urine. Results: After incorporation nerve agents quickly bind to proteins, e.g., acetylcholinesterase, butyrylcholinesterase or serum albumin, and only small parts remain freely circulating for a few hours (G-type) or up to 2 days (V-type). Concurrently they are converted to O-alkyl methylphosphonic acids by phosphotriesterases and/or simply by aqueous hydrolysis. As a re- sult, different biomarkers can be detected depending on the time passed between exposure and sampling. Unreacted V-type agents can be detected in plasma for 2 days, the O-alkyl methyl- phosphonic acids in plasma for about 2–4 days and in urine for up to 1 week. Fluoride-indu- ced release of protein-bound nerve agents can be carried out until 3 weeks post exposure. -

Warning: the Following Lecture Contains Graphic Images
What the новичок (Novichok)? Why Chemical Warfare Agents Are More Relevant Than Ever Matt Sztajnkrycer, MD PHD Professor of Emergency Medicine, Mayo Clinic Medical Toxicologist, Minnesota Poison Control System Medical Director, RFD Chemical Assessment Team @NoobieMatt #ITLS2018 Disclosures In accordance with the Accreditation Council for Continuing Medical Education (ACCME) Standards, the American Nurses Credentialing Center’s Commission (ANCC) and the Commission on Accreditation for Pre-Hospital Continuing Education (CAPCE), states presenters must disclose the existence of significant financial interests in or relationships with manufacturers or commercial products that may have a direct interest in the subject matter of the presentation, and relationships with the commercial supporter of this CME activity. The presenter does not consider that it will influence their presentation. Dr. Sztajnkrycer does not have a significant financial relationship to report. Dr. Sztajnkrycer is on the Editorial Board of International Trauma Life Support. Specific CW Agents Classes of Chemical Agents: The Big 5 The “A” List Pulmonary Agents Phosgene Oxime, Chlorine Vesicants Mustard, Phosgene Blood Agents CN Nerve Agents G, V, Novel, T Incapacitating Agents Thinking Outside the Box - An Abbreviated List Ammonia Fluorine Chlorine Acrylonitrile Hydrogen Sulfide Phosphine Methyl Isocyanate Dibotane Hydrogen Selenide Allyl Alcohol Sulfur Dioxide TDI Acrolein Nitric Acid Arsine Hydrazine Compound 1080/1081 Nitrogen Dioxide Tetramine (TETS) Ethylene Oxide Chlorine Leaks Phosphine Chlorine Common Toxic Industrial Chemical (“TIC”). Why use it in war/terror? Chlorine Density of 3.21 g/L. Heavier than air (1.28 g/L) sinks. Concentrates in low-lying areas. Like basements and underground bunkers. Reacts with water: Hypochlorous acid (HClO) Hydrochloric acid (HCl). -

Determination of Organophosphorus Pesticide Residues in Vegetables Using Solid Phase Micro-Extraction Coupled with Gas Chromatography–flame Photometric Detector
Arabian Journal of Chemistry (2015) xxx, xxx–xxx King Saud University Arabian Journal of Chemistry www.ksu.edu.sa www.sciencedirect.com ORIGINAL ARTICLE Determination of organophosphorus pesticide residues in vegetables using solid phase micro-extraction coupled with gas chromatography–flame photometric detector Haizarul Aida Sapahin, Ahmad Makahleh *, Bahruddin Saad * School of Chemical Sciences, Universiti Sains Malaysia, 11800 Minden, Penang, Malaysia Received 17 July 2014; accepted 9 December 2014 KEYWORDS Abstract An adequate and simple analytical method based on solid-phase microextraction Organophosphorus pesticide; (SPME) followed by gas chromatography–flame photometric detection (GC–FPD) for the determi- Direct immersed-solid phase nation of eleven organophosphorus pesticide residues (i.e., ethoprophos, sulfotep, diazinon, tolclo- microextraction; fos-methyl, fenitrothion, chlorpyrifos, isofenphos, methidathion, ethion, triazophos, leptophos) in Gas chromatography–flame vegetables samples (cabbage, kale and mustard) was developed. Important parameters that influ- photometric detector; ence the extraction efficiency (i.e., fibre type, extraction modes, extraction time, salt addition, Vegetables desorption time and temperature) were systematically investigated. Four types of commercially available fibres (i.e., 50/30 lm divinylbenzene/carboxen/polydimethylsiloxane (DVB/CAR/PDMS), 65 lm polydimethylsiloxane/divinylbenzene (PDMS/DVB), 100 lm polydimethylsiloxane (PDMS), and 85 lm polyacrylate (PA)) were evaluated. PA fibre exhibited the best performance and was used for the rest of the studies. The optimised extraction conditions were: extraction time, 30 min at room temperature; stirring speed, 1275 rpm; salt content, 10% NaCl; desorption time and temper- ature, 11 min at 260 °C; and no pH adjustment of the sample extract. The method was validated over the range 0.1–100 lg/L. -
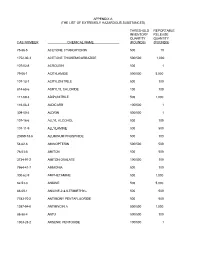
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

Malathion Human Health and Ecological Risk Assessment Final Report
SERA TR-052-02-02c Malathion Human Health and Ecological Risk Assessment Final Report Submitted to: Paul Mistretta, COR USDA/Forest Service, Southern Region 1720 Peachtree RD, NW Atlanta, Georgia 30309 USDA Forest Service Contract: AG-3187-C-06-0010 USDA Forest Order Number: AG-43ZP-D-06-0012 SERA Internal Task No. 52-02 Submitted by: Patrick R. Durkin Syracuse Environmental Research Associates, Inc. 5100 Highbridge St., 42C Fayetteville, New York 13066-0950 Fax: (315) 637-0445 E-Mail: [email protected] Home Page: www.sera-inc.com May 12, 2008 Table of Contents Table of Contents............................................................................................................................ ii List of Figures................................................................................................................................. v List of Tables ................................................................................................................................. vi List of Appendices ......................................................................................................................... vi List of Attachments........................................................................................................................ vi ACRONYMS, ABBREVIATIONS, AND SYMBOLS ............................................................... vii COMMON UNIT CONVERSIONS AND ABBREVIATIONS.................................................... x CONVERSION OF SCIENTIFIC NOTATION .......................................................................... -

744 Hydrolysis of Chiral Organophosphorus Compounds By
[Frontiers in Bioscience, Landmark, 26, 744-770, Jan 1, 2021] Hydrolysis of chiral organophosphorus compounds by phosphotriesterases and mammalian paraoxonase-1 Antonio Monroy-Noyola1, Damianys Almenares-Lopez2, Eugenio Vilanova Gisbert3 1Laboratorio de Neuroproteccion, Facultad de Farmacia, Universidad Autonoma del Estado de Morelos, Morelos, Mexico, 2Division de Ciencias Basicas e Ingenierias, Universidad Popular de la Chontalpa, H. Cardenas, Tabasco, Mexico, 3Instituto de Bioingenieria, Universidad Miguel Hernandez, Elche, Alicante, Spain TABLE OF CONTENTS 1. Abstract 2. Introduction 2.1. Organophosphorus compounds (OPs) and their toxicity 2.2. Metabolism and treatment of OP intoxication 2.3. Chiral OPs 3. Stereoselective hydrolysis 3.1. Stereoselective hydrolysis determines the toxicity of chiral compounds 3.2. Hydrolysis of nerve agents by PTEs 3.2.1. Hydrolysis of V-type agents 3.3. PON1, a protein restricted in its ability to hydrolyze chiral OPs 3.4. Toxicity and stereoselective hydrolysis of OPs in animal tissues 3.4.1. The calcium-dependent stereoselective activity of OPs associated with PON1 3.4.2. Stereoselective hydrolysis commercial OPs pesticides by alloforms of PON1 Q192R 3.4.3. PON1, an enzyme that stereoselectively hydrolyzes OP nerve agents 3.4.4. PON1 recombinants and stereoselective hydrolysis of OP nerve agents 3.5. The activity of PTEs in birds 4. Conclusions 5. Acknowledgments 6. References 1. ABSTRACT Some organophosphorus compounds interaction of the racemic OPs with these B- (OPs), which are used in the manufacturing of esterases (AChE and NTE) and such interactions insecticides and nerve agents, are racemic mixtures have been studied in vivo, ex vivo and in vitro, using with at least one chiral center with a phosphorus stereoselective hydrolysis by A-esterases or atom. -

Chemical Warfare Agents
Manuscript for Kirk-Othmer Encyclopedia of Chemical Technology August 2019 CHEMICAL WARFARE AGENTS This is the pre-print manuscript of an article published in the Kirk-Othmer Encyclopedia of Chemical Technology: https://onlinelibrary.wiley.com/doi/book/10.1002/0471238961 The published version of the article is available at the Wiley website: https://onlinelibrary.wiley.com/doi/10.1002/0471238961.0308051308011818.a01.pub3 How to cite: Costanzi, S. (2020). Chemical Warfare Agents. In Kirk‐Othmer Encyclopedia of Chemical Technology, (Ed.). doi:10.1002/0471238961.0308051308011818.a01.pub3 Stefano Costanzi Department of Chemistry and Center for Behavioral Neuroscience American University, Washington, D.C. [email protected] Chemical weapons are weapons that exploit the toxicity of chemicals to bring about death or harm. The toxic chemicals on which chemical weapons are based are known as chemical warfare agents. The elimination of this entire category of weapons is the aim of the Convention on the Prohibition of the Development, Production, Stockpiling and Use of Chemical Weapons and on their Destruction, also known as Chemical Weapons Convention or CWC, which was opened for signature in 1993 and entered into force in 1997. Administered and implemented by the Hague- based Organisation for the Prohibition of Chemical Weapons (OPCW), the CWC is an international treaty that enjoys almost universal embracement, having been ratified or acceded by 193 States Parties. Importantly, the CWC poses a complete and absolute ban on chemical weapons, mandating State Parties to renounce “(a) to develop, produce, otherwise acquire, stockpile or retain chemical weapons, or transfer, directly or indirectly, chemical weapons to anyone; (b) to use chemical weapons; (c) to engage in any military preparations to use chemical weapons; (d) to assist, encourage or induce, in any way, anyone to engage in any activity prohibited to a State Party” under the Convention (CWC Article II, Paragraph 1) (1-3). -

Environmental Health Criteria 63 ORGANOPHOSPHORUS
Environmental Health Criteria 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION Please note that the layout and pagination of this web version are not identical with the printed version. Organophophorus insecticides: a general introduction (EHC 63, 1986) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1986 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination -

Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 Theinternational Programme on Chemical Safety (IPCS) Was Established in 1980
The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 cation Hazard of Pesticides by and Guidelines to Classi The WHO Recommended Classi The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 TheInternational Programme on Chemical Safety (IPCS) was established in 1980. The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. This publication was developed in the IOMC context. The contents do not necessarily reflect the views or stated policies of individual IOMC Participating Organizations. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase international coordination in the field of chemical safety. The Participating Organizations are: FAO, ILO, UNDP, UNEP, UNIDO, UNITAR, WHO, World Bank and OECD. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment. WHO recommended classification of pesticides by hazard and guidelines to classification, 2019 edition ISBN 978-92-4-000566-2 (electronic version) ISBN 978-92-4-000567-9 (print version) ISSN 1684-1042 © World Health Organization 2020 Some rights reserved. -

The Benefit of Combinations of Oximes for the Ability of Antidotal Treatment
Caisberger et al. BMC Pharmacology and Toxicology (2018) 19:35 https://doi.org/10.1186/s40360-018-0227-0 RESEARCH ARTICLE Open Access The benefit of combinations of oximes for the ability of antidotal treatment to counteract sarin-induced brain damage in rats Filip Caisberger1, Jaroslav Pejchal2, Jan Misik2, Jiri Kassa2, Martin Valis1 and Kamil Kuca3,4* Abstract Background: The aim of our study was to compare the ability of two combinations of oximes (HI-6 + trimedoxime and HI-6 + K203) with atropine to counteract acute sarin-induced brain damage with the efficacy of antidotal treatment involving single oxime (HI-6) and atropin using in vivo methods. Methods: Brain damage and neuroprotective effects of antidotal treatment were evaluated in rats poisoned with sarin at a sublethal dose (108 μg/kg i.m.; 90% LD50) using histopathological, Fluoro-Jade B and Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) analysis 24 h after sarin administration. Results: Both combinations of oximes reduce the number of rats that died before the end of experiment compared to non-treated sarin poisoning and sarin poisoning treated with HI-6 and atropine. In the case of treatment of sarin poisoning with HI-6 in combination with K203, all rats survived till the end of experiment. HI-6 with atropine was able to reduce sarin-induced brain damage, however, both combinations were slightly more effective. Conclusions: The oxime HI-6 in combination with K203 and atropine seems to be the most effective. Thus, both tested oxime combinations bring a small benefit in elimination of acute sarin-induced brain damage compared to single oxime antidotal therapy. -

Carbaryl Human Health and Ecological Risk Assessment Revised Final Report
SERA TR-052-01-05a Carbaryl Human Health and Ecological Risk Assessment Revised Final Report Submitted to: Paul Mistretta, COR USDA/Forest Service, Southern Region 1720 Peachtree RD, NW Atlanta, Georgia 30309 USDA Forest Service Contract: AG-3187-C-06-0010 USDA Forest Order Number: AG-43ZP-D-06-0009 SERA Internal Task No. 52-01 Submitted by: Patrick R. Durkin and Cynthia King Syracuse Environmental Research Associates, Inc. 5100 Highbridge St., 42C Fayetteville, New York 13066-0950 Fax: (315) 637-0445 E-Mail: [email protected] Home Page: www.sera-inc.com February 9, 2008 Table of Contents Table of Contents............................................................................................................................ ii List of Figures................................................................................................................................. v List of Tables .................................................................................................................................. v List of Attachments........................................................................................................................ vi List of Appendices ......................................................................................................................... vi COMMON UNIT CONVERSIONS AND ABBREVIATIONS................................................... ix CONVERSION OF SCIENTIFIC NOTATION ............................................................................ x EXECUTIVE SUMMARY ..........................................................................................................