Determination of Semivolatile Organic Compounds and Polycyclic Aromatic Hydrocarbons in Solids by Gas Chromatography/Mass Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polycyclic Aromatic Hydrocarbon Migration from Creosote-Treated Railway Ties Into Ballast and Adjacent Wetlands
United States In cooperation Department of with the Agriculture Polycyclic Aromatic United States Forest Service Department of Transportation Hydrocarbon Migration Forest Products Federal Laboratory Highway Administration From Creosote-Treated Research Paper FPL−RP−617 Railway Ties Into Ballast and Adjacent Wetlands Kenneth M. Brooks Abstract from the weathered ties at this time. No significant PAH loss was observed from ties during the second summer. A Occasionally, creosote-treated railroad ties need to be small portion of PAH appeared to move vertically down into replaced, sometimes in sensitive environments such as the ballast to approximately 60 cm. Small amounts of PAH wetlands. To help determine if this is detrimental to the may have migrated from the ballast into adjacent wetlands surrounding environment, more information is needed on during the second summer, but these amounts were not the extent and pattern of creosote, or more specifically poly- statistically significant. These results suggest that it is rea- cyclic aromatic hydrocarbon (PAH), migration from railroad sonable to expect a detectable migration of creosote-derived ties and what effects this would have on the surrounding PAH from newly treated railway ties into supporting ballast environment. This study is a report on PAH level testing during their first exposure to hot summer weather. The PAH done in a simulated wetland mesocosm. Both newly treated rapidly disappeared from the ballast during the fall and and weathered creosote-treated railroad ties were placed in winter following this initial loss. Then statistically insignifi- the simulated wetland. As a control, untreated ties were also cant vertical and horizontal migration of these PAH suggests placed in the mesocosm. -

Safeguards Benzopyrene Contamination in Korean
SAFEGUARDS SGS CONSUMER TESTING SERVICES FOOD NO. 187/12 NOV 2012 BENZOPYRENE CONTAMINATION IN KOREAN INSTANT NOODLE On 25 Oct 2012, Korea Food and Drug Administration (KFDA) recalled six instant noodle products from the nation’s largest ramen maker in South Korea because these products were found to be tainted with benzo(a) pyrene, which is a known carcinogen.1 Several countries, including Philippines, Thailand, Taiwan and China, have been alerted to this and as a precaution, have removed these products from their shelves. Benzo(a)pyrene belongs to the group of polycyclic aromatic hydrocarbons (PAHs). Most PAHs derive from incomplete burning of carbon containing materials such as petroleum fuels, wood, and garbage. The occurrence of PAHs in foods is mainly caused by environmental contamination (water, air and soil), food processing (direct drying and heating), and packaging materials. PAHs have been found in different food products, such as dairy products, vegetables, fruits, oil, coffee, tea, cereals, smoked meat, seafood products, ingredients for food formulation, seasoning, and instant noodles. announcement of the test results and the recall order because the levels of In June, the KFDA conducted tests on benzopyrene found in the noodle products in June were minuscule and not harmful, 30 instant noodle products sold in the South Korean agency reported. However, the KFDA issued the recall order after South Korea and found that six its commissioner Lee Hee-sung was pressed on the matter during a legislative products contained benzopyrene, with question-and-answer, the report indicated.2 one product containing the highest level of 4.7 parts per billion (µg/kg). -

Polycyclic Aromatic Hydrocarbon Structure Index
NIST Special Publication 922 Polycyclic Aromatic Hydrocarbon Structure Index Lane C. Sander and Stephen A. Wise Chemical Science and Technology Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899-0001 December 1997 revised August 2020 U.S. Department of Commerce William M. Daley, Secretary Technology Administration Gary R. Bachula, Acting Under Secretary for Technology National Institute of Standards and Technology Raymond G. Kammer, Director Polycyclic Aromatic Hydrocarbon Structure Index Lane C. Sander and Stephen A. Wise Chemical Science and Technology Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899 This tabulation is presented as an aid in the identification of the chemical structures of polycyclic aromatic hydrocarbons (PAHs). The Structure Index consists of two parts: (1) a cross index of named PAHs listed in alphabetical order, and (2) chemical structures including ring numbering, name(s), Chemical Abstract Service (CAS) Registry numbers, chemical formulas, molecular weights, and length-to-breadth ratios (L/B) and shape descriptors of PAHs listed in order of increasing molecular weight. Where possible, synonyms (including those employing alternate and/or obsolete naming conventions) have been included. Synonyms used in the Structure Index were compiled from a variety of sources including “Polynuclear Aromatic Hydrocarbons Nomenclature Guide,” by Loening, et al. [1], “Analytical Chemistry of Polycyclic Aromatic Compounds,” by Lee et al. [2], “Calculated Molecular Properties of Polycyclic Aromatic Hydrocarbons,” by Hites and Simonsick [3], “Handbook of Polycyclic Hydrocarbons,” by J. R. Dias [4], “The Ring Index,” by Patterson and Capell [5], “CAS 12th Collective Index,” [6] and “Aldrich Structure Index” [7]. In this publication the IUPAC preferred name is shown in large or bold type. -

Estimation of Enthalpy of Bio-Oil Vapor and Heat Required For
Edinburgh Research Explorer Estimation of Enthalpy of Bio-Oil Vapor and Heat Required for Pyrolysis of Biomass Citation for published version: Yang, H, Kudo, S, Kuo, H-P, Norinaga, K, Mori, A, Masek, O & Hayashi, J 2013, 'Estimation of Enthalpy of Bio-Oil Vapor and Heat Required for Pyrolysis of Biomass', Energy & Fuels, vol. 27, no. 5, pp. 2675-2686. https://doi.org/10.1021/ef400199z Digital Object Identifier (DOI): 10.1021/ef400199z Link: Link to publication record in Edinburgh Research Explorer Document Version: Early version, also known as pre-print Published In: Energy & Fuels General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 04. Oct. 2021 Estimation of Enthalpy of Bio-oil Vapor and Heat Required for Pyrolysis of Biomass Hua Yang,† Shinji Kudo,§ Hsiu-Po Kuo,‡ Koyo Norinaga, ξ Aska Mori, ξ Ondřej Mašek,|| and Jun-ichiro Hayashi †, ξ, §, * †Interdisciplinary Graduate School of Engineering Sciences, Kyushu University, -

Analysis of Benzopyrenes and Benzopyrene Metabolites by Fluorescence Spectroscopy Techniques
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2016 Analysis of Benzopyrenes and Benzopyrene Metabolites by Fluorescence Spectroscopy Techniques Bassam Al-Farhani University of Central Florida Part of the Chemistry Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Doctoral Dissertation (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Al-Farhani, Bassam, "Analysis of Benzopyrenes and Benzopyrene Metabolites by Fluorescence Spectroscopy Techniques" (2016). Electronic Theses and Dissertations, 2004-2019. 5286. https://stars.library.ucf.edu/etd/5286 ANALYSIS OF BENZOPYRENES AND BENZOPYRENE METABOLITES BY FLUORESCENCE SPECTROSCOPY TECHNIQUES by BASSAM AL-FARHANI B.Sc. Alnahrain University Iraq, 2002 M.Sc. Alnahrain University Iraq, 2005 A dissertation submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry in the College of Sciences at the University of Central Florida Orlando, Florida Spring Term 2016 Major Professor: Andres D. Campiglia © 2016 Bassam Al-farhani ii ABSTRACT Polycyclic aromatic hydrocarbons (PAHs) are some of the most common and toxic pollutants encountered worldwide. Presently, monitoring is restricted to sixteen PAHs, but it is well understood that this list omits many toxic PAHs. Among the “forgotten” PAHs, isomers with molecular weight 302 are of particular concern due to their high toxicological properties. The chromatographic analysis of PAHs with MW 302 is challenged by similar retention times and virtually identical mass fragmentation patterns. -

Ambient Water Quality Criteria for Polycyclic Aromatic Hydrocarbons (Pahs)
Ambient Water Quality Criteria For Polycyclic Aromatic Hydrocarbons (PAHs) Ministry of Environment, Lands and Parks Province of British Columbia N. K. Nagpal, Ph.D. Water Quality Branch Water Management Division February, 1993 ACKNOWLEDGEMENTS The author is indebted to the following individual and agencies for providing valuable comments during the preparation of this document. Dr. Ray Copes BC. Ministry of Health, Victoria, BC. Dr. G. R. Fox Environmental Protection Div., BC. MOELP, Victoria, BC. Mr. L. W. Pommen Water Quality Branch, BC. MOELP, Victoria, BC. Mr. R. J. Rocchini Water Quality Branch, BC. MOELP, Victoria, BC. Ms. Sherry Smith Eco-Health Branch, Conservation and Protection, Environment Canada, Hull, Quebec Mr. Scott Teed Eco-Health Branch, Conservation and Protection, Environment Canada, Hull, Quebec Ms. Bev Raymond Integrated Programs Branch, Inland Waters, Environment Canada, North Vancouver, BC. 1.0 INTRODUCTION Polycyclic aromatic hydrocarbons (PAHs) are organic compounds which are non- essential for the growth of plants, animals or humans; yet, they are ubiquitous in the environment. When present in sufficient quantity in the environment, certain PAHs are toxic and carcinogenic to plants, animals and humans. This document discusses the characteristics of PAHs and their effects on various water uses, which include drinking Ministry of Environment Water Protection and Sustainability Branch Mailing Address: Telephone: 250 387-9481 Environmental Sustainability PO Box 9362 Facsimile: 250 356-1202 and Strategic Policy Division Stn Prov Govt Website: www.gov.bc.ca/water Victoria BC V8W 9M2 water, aquatic life, wildlife, livestock watering, irrigation, recreation and aesthetics, and industrial water supplies. A significant portion of this document discusses the effects of PAHs upon aquatic life, due to its sensitivity to PAHs. -

Summary of Information for ABC for Polycyclic Aromatic Hydrocarbons
Air Toxics Science Advisory Committee Summary of Information for ABC for Polycyclic Aromatic Hydrocarbons September 16, 2015 ATSAC Meeting #10 Presenter: Sue MacMillan, DEQ ATSAC lead Sue MacMillan | Oregon Department of Environmental Quality 26 Individual PAHs to Serve as Basis of ABC for Total PAHs Acenaphthene Cyclopenta(c,d)pyrene Acenaphthylene Dibenzo(a,h)anthracene Anthracene Dibenzo(a,e)pyrene Anthanthrene Dibenzo(a,h)pyrene Benzo(a)pyrene Dibenzo(a,i)pyrene Naphthalene Benz(a)anthracene Dibenzo(a,l)pyrene has separate Benzo(b)fluoranthene Fluoranthene ABC. Benzo(k)fluoranthene Fluorene Benzo( c)pyrene Indeno(1,2,3-c,d)pyrene Benzo(e)pyrene Phenanthrene Benzo(g,h,i)perylene Pyrene Benzo(j)fluoranthene 5-Methylchrysene Chrysene 6-Nitrochrysene Use of Toxic Equivalency Factors for PAHs • Benzo(a)pyrene serves as the index PAH, and has a documented toxicity value to which other PAHs are adjusted • Other PAHs adjusted using Toxic Equivalency Factors (TEFs), aka Potency Equivalency Factors (PEFs). These values are multipliers and are PAH-specific. • Once all PAH concentrations are adjusted to account for their relative toxicity as compared to BaP, the concentrations are summed • This summed concentration is then compared to the toxicity value for BaP, which is used as the ABC for total PAHs. Source of PEFs for PAHs • EPA provides a range of values of PEFs for each PAH • Original proposal suggested using upper-bound value of each PEF range as the PEF to use for adjustment of our PAHs • Average PEF value for each PAH is a better approximation of central tendency, and is consistent with the use of PEFs by other agencies • Result of using average, rather than upper-bound PEFs: slightly lower summed concentrations for adjusted PAHs, thus less apt to exceed ABC for total PAHs Documents can be provided upon request in an alternate format for individuals with disabilities or in a language other than English for people with limited English skills. -
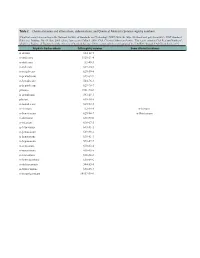
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

Polycyclic Aromatic Hydrocarbons (Pahs)
Polycyclic Aromatic Hydrocarbons (PAHs) Factsheet 4th edition Donata Lerda JRC 66955 - 2011 The mission of the JRC-IRMM is to promote a common and reliable European measurement system in support of EU policies. European Commission Joint Research Centre Institute for Reference Materials and Measurements Contact information Address: Retiewseweg 111, 2440 Geel, Belgium E-mail: [email protected] Tel.: +32 (0)14 571 826 Fax: +32 (0)14 571 783 http://irmm.jrc.ec.europa.eu/ http://www.jrc.ec.europa.eu/ Legal Notice Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use which might be made of this publication. Europe Direct is a service to help you find answers to your questions about the European Union Freephone number (*): 00 800 6 7 8 9 10 11 (*) Certain mobile telephone operators do not allow access to 00 800 numbers or these calls may be billed. A great deal of additional information on the European Union is available on the Internet. It can be accessed through the Europa server http://europa.eu/ JRC 66955 © European Union, 2011 Reproduction is authorised provided the source is acknowledged Printed in Belgium Table of contents Chemical structure of PAHs................................................................................................................................. 1 PAHs included in EU legislation.......................................................................................................................... 6 Toxicity of PAHs included in EPA and EU -

Health Risks of Structural Firefighters from Exposure to Polycyclic
International Journal of Environmental Research and Public Health Systematic Review Health Risks of Structural Firefighters from Exposure to Polycyclic Aromatic Hydrocarbons: A Systematic Review and Meta-Analysis Jooyeon Hwang 1,* , Chao Xu 2 , Robert J. Agnew 3 , Shari Clifton 4 and Tara R. Malone 4 1 Department of Occupational and Environmental Health, Hudson College of Public Health, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA 2 Department of Biostatistics and Epidemiology, Hudson College of Public Health, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA; [email protected] 3 Fire Protection & Safety Engineering Technology Program, College of Engineering, Architecture and Technology, Oklahoma State University, Stillwater, OK 74078, USA; [email protected] 4 Department of Health Sciences Library and Information Management, Graduate College, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA; [email protected] (S.C.); [email protected] (T.R.M.) * Correspondence: [email protected]; Tel.: +1-405-271-2070 (ext. 40415) Abstract: Firefighters have an elevated risk of cancer, which is suspected to be caused by occupational and environmental exposure to fire smoke. Among many substances from fire smoke contaminants, one potential source of toxic exposure is polycyclic aromatic hydrocarbons (PAH). The goal of this paper is to identify the association between PAH exposure levels and contributing risk factors to Citation: Hwang, J.; Xu, C.; Agnew, derive best estimates of the effects of exposure on structural firefighters’ working environment in R.J.; Clifton, S.; Malone, T.R. Health fire. We surveyed four databases (Embase, Medline, Scopus, and Web of Science) for this systematic Risks of Structural Firefighters from literature review. -

Ocular Effects from Burn Pits in Iraq, Afghanistan, and the Horn of Africa
Ocular effects from Burn Pits in Iraq, Afghanistan, and the Horn of Africa **NOTICE: this is not official VA or C&P released information, this is just information for you to use in assessing patients with exposure to burn pits compiled by Makesha Sink, OD*** Information on burn pits: - Large burn pits have been used throughout the operations in Iraq and Afghanistan to dispose of nearly all forms of waste. It is estimated that such pits, some nearly as large as 20 acres, are or have been located at every military forward operating base (FOB). The pit at Joint Base Balad, also known as Logistic Support Area (LSA) Anaconda, has received the most attention. The burned waste products include, but are not limited to: plastics, metal/aluminum cans, rubber, chemicals (such as, paints, solvents), petroleum and lubricant products, munitions and other unexploded ordnance, wood waste, medical and human waste, and incomplete combustion by- products. Jet fuel (JP-8) is used as the accelerant. The pits do not effectively burn the volume of waste generated, and smoke from the burn pit blows over bases and into living areas. - DoD has performed air sampling at Joint Base Balad, Iraq and Camp Lemonier, Djibouti. Subsequently, DoD has indicated that most of the air samples have not shown individual chemicals that exceed military exposure guidelines (MEG). Nonetheless, DoD further concluded that the confidence level in their risk estimates is low to medium due to lack of specific exposure information, other routes/sources of environmental hazards not identified; and uncertainty regarding the synergistic impact of multiple chemicals present, particularly those affecting the same body organs/systems. -

Fluoranthene
) , "- United States Office of Water EPA 440/5-80-049 Environmental Protection Regulations and Standards October 1980 Agency Criteria and Standards Division Washington DC 20460 aEPA Ambient Water Quality Criteria for Fluoranthene ... AMBIENT WATER QUALITY CRITERIA FOR FLUORANTHENE Prepared By U.S. ENVIRONMENTAL PROTECTION AGENCY Office of Water Regulations and Standards Criteria and Standards Division Washington, D.C. Office of Research and Development Environmental Criteria and Assessment Office Cincinnati, Ohio Carcinogen Assessment Group Washington, D.C. Environmental Research Laboratories Corvalis, Oregon Duluth, Minnesota Gulf Breeze, Florida Narragansett, Rhode Island - -J i DISCLAIMER This report has been reviewed by the Environmental Criteria and Assessment Office, U.S. Environmental Protection Agency, and approved for publication. Mention of trade names or commercial products does not constitute endorsement or recommendation for use. AVAILABILITY NOTICE This document is available to the public through the National Technical Information Service, (NTIS), Springfield, Virginia 22161. ii FOREWORD Section 304 (a) (1) of the Clean Water Act of 1977 (P.L. 95-217), requires the Administrator of the Environmental Protection Agency to publish criteria for water quality accurately reflecting the latest scientific knowledge on the kind and extent of all identifiable effects on health and we 1fare wh ich may be expected from the presence of pollutants in any body of water, including ground water. Proposed water quality criteria for the 65 toxic pollutants listed under section 307 (a) (1) of the Cl ean Water Act were deve loped and a not ice of their availability was published for public comment on March 15, 1979 (44 FR 15926), July 25, 1979 (44 FR 43660), and October 1, 1979 (44 FR 56628).