Analysis of Benzopyrenes and Benzopyrene Metabolites by Fluorescence Spectroscopy Techniques
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polycyclic Aromatic Hydrocarbon Migration from Creosote-Treated Railway Ties Into Ballast and Adjacent Wetlands
United States In cooperation Department of with the Agriculture Polycyclic Aromatic United States Forest Service Department of Transportation Hydrocarbon Migration Forest Products Federal Laboratory Highway Administration From Creosote-Treated Research Paper FPL−RP−617 Railway Ties Into Ballast and Adjacent Wetlands Kenneth M. Brooks Abstract from the weathered ties at this time. No significant PAH loss was observed from ties during the second summer. A Occasionally, creosote-treated railroad ties need to be small portion of PAH appeared to move vertically down into replaced, sometimes in sensitive environments such as the ballast to approximately 60 cm. Small amounts of PAH wetlands. To help determine if this is detrimental to the may have migrated from the ballast into adjacent wetlands surrounding environment, more information is needed on during the second summer, but these amounts were not the extent and pattern of creosote, or more specifically poly- statistically significant. These results suggest that it is rea- cyclic aromatic hydrocarbon (PAH), migration from railroad sonable to expect a detectable migration of creosote-derived ties and what effects this would have on the surrounding PAH from newly treated railway ties into supporting ballast environment. This study is a report on PAH level testing during their first exposure to hot summer weather. The PAH done in a simulated wetland mesocosm. Both newly treated rapidly disappeared from the ballast during the fall and and weathered creosote-treated railroad ties were placed in winter following this initial loss. Then statistically insignifi- the simulated wetland. As a control, untreated ties were also cant vertical and horizontal migration of these PAH suggests placed in the mesocosm. -

Safeguards Benzopyrene Contamination in Korean
SAFEGUARDS SGS CONSUMER TESTING SERVICES FOOD NO. 187/12 NOV 2012 BENZOPYRENE CONTAMINATION IN KOREAN INSTANT NOODLE On 25 Oct 2012, Korea Food and Drug Administration (KFDA) recalled six instant noodle products from the nation’s largest ramen maker in South Korea because these products were found to be tainted with benzo(a) pyrene, which is a known carcinogen.1 Several countries, including Philippines, Thailand, Taiwan and China, have been alerted to this and as a precaution, have removed these products from their shelves. Benzo(a)pyrene belongs to the group of polycyclic aromatic hydrocarbons (PAHs). Most PAHs derive from incomplete burning of carbon containing materials such as petroleum fuels, wood, and garbage. The occurrence of PAHs in foods is mainly caused by environmental contamination (water, air and soil), food processing (direct drying and heating), and packaging materials. PAHs have been found in different food products, such as dairy products, vegetables, fruits, oil, coffee, tea, cereals, smoked meat, seafood products, ingredients for food formulation, seasoning, and instant noodles. announcement of the test results and the recall order because the levels of In June, the KFDA conducted tests on benzopyrene found in the noodle products in June were minuscule and not harmful, 30 instant noodle products sold in the South Korean agency reported. However, the KFDA issued the recall order after South Korea and found that six its commissioner Lee Hee-sung was pressed on the matter during a legislative products contained benzopyrene, with question-and-answer, the report indicated.2 one product containing the highest level of 4.7 parts per billion (µg/kg). -

Polycyclic Aromatic Hydrocarbon Structure Index
NIST Special Publication 922 Polycyclic Aromatic Hydrocarbon Structure Index Lane C. Sander and Stephen A. Wise Chemical Science and Technology Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899-0001 December 1997 revised August 2020 U.S. Department of Commerce William M. Daley, Secretary Technology Administration Gary R. Bachula, Acting Under Secretary for Technology National Institute of Standards and Technology Raymond G. Kammer, Director Polycyclic Aromatic Hydrocarbon Structure Index Lane C. Sander and Stephen A. Wise Chemical Science and Technology Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899 This tabulation is presented as an aid in the identification of the chemical structures of polycyclic aromatic hydrocarbons (PAHs). The Structure Index consists of two parts: (1) a cross index of named PAHs listed in alphabetical order, and (2) chemical structures including ring numbering, name(s), Chemical Abstract Service (CAS) Registry numbers, chemical formulas, molecular weights, and length-to-breadth ratios (L/B) and shape descriptors of PAHs listed in order of increasing molecular weight. Where possible, synonyms (including those employing alternate and/or obsolete naming conventions) have been included. Synonyms used in the Structure Index were compiled from a variety of sources including “Polynuclear Aromatic Hydrocarbons Nomenclature Guide,” by Loening, et al. [1], “Analytical Chemistry of Polycyclic Aromatic Compounds,” by Lee et al. [2], “Calculated Molecular Properties of Polycyclic Aromatic Hydrocarbons,” by Hites and Simonsick [3], “Handbook of Polycyclic Hydrocarbons,” by J. R. Dias [4], “The Ring Index,” by Patterson and Capell [5], “CAS 12th Collective Index,” [6] and “Aldrich Structure Index” [7]. In this publication the IUPAC preferred name is shown in large or bold type. -

Ambient Water Quality Criteria for Polycyclic Aromatic Hydrocarbons (Pahs)
Ambient Water Quality Criteria For Polycyclic Aromatic Hydrocarbons (PAHs) Ministry of Environment, Lands and Parks Province of British Columbia N. K. Nagpal, Ph.D. Water Quality Branch Water Management Division February, 1993 ACKNOWLEDGEMENTS The author is indebted to the following individual and agencies for providing valuable comments during the preparation of this document. Dr. Ray Copes BC. Ministry of Health, Victoria, BC. Dr. G. R. Fox Environmental Protection Div., BC. MOELP, Victoria, BC. Mr. L. W. Pommen Water Quality Branch, BC. MOELP, Victoria, BC. Mr. R. J. Rocchini Water Quality Branch, BC. MOELP, Victoria, BC. Ms. Sherry Smith Eco-Health Branch, Conservation and Protection, Environment Canada, Hull, Quebec Mr. Scott Teed Eco-Health Branch, Conservation and Protection, Environment Canada, Hull, Quebec Ms. Bev Raymond Integrated Programs Branch, Inland Waters, Environment Canada, North Vancouver, BC. 1.0 INTRODUCTION Polycyclic aromatic hydrocarbons (PAHs) are organic compounds which are non- essential for the growth of plants, animals or humans; yet, they are ubiquitous in the environment. When present in sufficient quantity in the environment, certain PAHs are toxic and carcinogenic to plants, animals and humans. This document discusses the characteristics of PAHs and their effects on various water uses, which include drinking Ministry of Environment Water Protection and Sustainability Branch Mailing Address: Telephone: 250 387-9481 Environmental Sustainability PO Box 9362 Facsimile: 250 356-1202 and Strategic Policy Division Stn Prov Govt Website: www.gov.bc.ca/water Victoria BC V8W 9M2 water, aquatic life, wildlife, livestock watering, irrigation, recreation and aesthetics, and industrial water supplies. A significant portion of this document discusses the effects of PAHs upon aquatic life, due to its sensitivity to PAHs. -
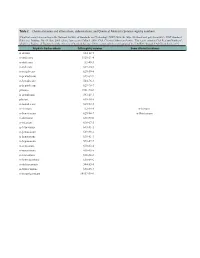
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

Ambient Air Pollution by Polycyclic Aromatic Hydrocarbons (PAH)
Ambient Air Pollution by Polycyclic Aromatic Hydrocarbons (PAH) Position Paper Annexes July 27th 2001 Prepared by the Working Group On Polycyclic Aromatic Hydrocarbons PAH Position Paper Annexes July 27th 2001 i PAH Position Paper Annexes July 27th 2001 Contents ANNEX 1...............................................................................................................................................................1 MEMBERSHIP OF THE WORKING GROUP .............................................................................................................1 ANNEX 2...............................................................................................................................................................3 Tables and Figures 3 Table 1: Physical Properties and Structures of Selected PAH 4 Table 2: Details of carcinogenic groups and measurement lists of PAH 9 Table 3: Review of Legislation or Guidance intended to limit ambient air concentrations of PAH. 10 Table 4: Emissions estimates from European countries - Anthropogenic emissions of PAH (tonnes/year) in the ECE region 12 Table 5: Summary of recent (not older than 1990) typical European PAH- and B(a)P concentrations in ng/m3 as annual mean value. 14 Table 6: Summary of benzo[a]pyrene Emissions in the UK 1990-2010 16 Table 7: Current network designs at national level (end-1999) 17 Table 8: PAH sampling and analysis methods used in several European countries. 19 Table 9: BaP collected as vapour phase in European investigations: percent relative to total (vapour -

7 Benzene and Aromatics
Benzene and Aromatic Compounds Chapter 15 Organic Chemistry, 8th Edition John McMurry 1 Background • Benzene (C6H6) is the simplest aromatic hydrocarbon (or arene). • Four degrees of unsaturation. • It is planar. • All C—C bond lengths are equal. • Whereas unsaturated hydrocarbons such as alkenes, alkynes and dienes readily undergo addition reactions, benzene does not. • Benzene reacts with bromine only in the presence of FeBr3 (a Lewis acid), and the reaction is a substitution, not an addition. 2 The Structure of Benzene: MO 3 The Structure of Benzene: Resonance The true structure of benzene is a resonance hybrid of the two Lewis structures. 4 Aromaticity – Resonance Energy 5 Stability of Benzene - Aromaticity Benzene does not undergo addition reactions typical of other highly unsaturated compounds, including conjugated dienes. 6 The Criteria for Aromaticity Four structural criteria must be satisfied for a compound to be aromatic. [1] A molecule must be cyclic. 7 The Criteria for Aromaticity [2] A molecule must be completely conjugated (all atoms sp2). 8 The Criteria for Aromaticity [3] A molecule must be planar. 9 The Criteria for Aromaticity—Hückel’s Rule [4] A molecule must satisfy Hückel’s rule. 10 The Criteria for Aromaticity—Hückel’s Rule 1. Aromatic—A cyclic, planar, completely conjugated compound with 4n + 2 π electrons. 3. Antiaromatic—A cyclic, planar, completely conjugated compound with 4n π electrons. 5. Not aromatic (nonaromatic)—A compound that lacks one (or more) of the following requirements for aromaticity: being cyclic, -

1. Introduction
THE ASTROPHYSICAL JOURNAL, 502:284È295, 1998 July 20 ( 1998. The American Astronomical Society. All rights reserved. Printed in U.S.A. INDIGENOUS POLYCYCLIC AROMATIC HYDROCARBONS IN CIRCUMSTELLAR GRAPHITE GRAINS FROM PRIMITIVE METEORITES S.MESSENGER,1 S.AMARI,W X. GAO, AND R. M. ALKER McDonnell Center for the Space Sciences and Physics Department, Washington University, One Brookings Drive, St. Louis, MO 63130; srm=nist.gov, sa=howdy.wustl.edu, xg=howdy.wustl.edu, rmw=howdy.wustl.edu S. J.CLEMETT,C X. D. F. HILLIER,2 AND R. N. ZARE Department of Chemistry, Stanford University, Stanford, CA 94305; simon=d31rz3.stanford.edu, chillier=ssa1.arc.nasa.gov, rnz=d31rz3.stanford.edu AND R. S. LEWIS Enrico Fermi Institute, University of Chicago, 5630 Ellis Avenue, Chicago, IL 60637; royl=rainbow.uchicago.edu Received 1997 May 21; accepted 1998 March 4 ABSTRACT We report the measurement of polycyclic aromatic hydrocarbons (PAHs) in individual circumstellar graphite grains extracted from two primitive meteorites, Murchison and Acfer 094. The 12C/13C isotope ratios of the grains in this study range from 2.4 to 1700. Roughly 70% of the grains have an appreciable concentration of PAHs (500È5000 parts per million [ppm]). Independent molecule-speciÐc isotopic analyses show that most of the PAHs appear isotopically normal, but in several cases correlated isotopic anomalies are observed between one or more molecules and their parent grains. These correlations are most evident for 13C-depleted grains. Possible origins of the PAHs in the graphite grains are discussed. Subject headings: circumstellar matter È dust, extinction È ISM: molecules È molecular processes È stars: carbon 1. -

Health Impact of Corn Ethanol Blends
Health Impact of Corn Ethanol Blends NCGA Meeting St. Louis, MO Steffen Mueller, PhD, Principal Economist December 2018 Study Contributors Authors • Steffen Mueller, Principal Economist, University of Illinois at Chicago, Energy Resources Center. • Stefan Unnasch, Managing Director, Life Cycle Associates, LLC. • Bill Keesom, Retired UOP Refinery and Fuels Consultant, Evanston, Illinois\ • Samartha Mohan, Engineer, Automotive Technology, Bangalor, India • Love Goyal, Analyst, Life Cycle Associates • Professor Rachael Jones; University of Illinois at Chicago School of Public Health Technical Input and/or Review: • Dr. Zigang Dong, Executive Director, and Dr. K S Reddy, The Hormel Institute, University of Minnesota. • Brian West, John Storey, Shean Huff, John Thomas; Fuels, Engines, and Emissions Research Center, Oak Ridge National Laboratory • Professor Jane Lin; University of Illinois at Chicago. Department of Civil Engineering. • Kristi Moore; Fueltech Service. • Jordan Rockensuess, Intertek Laboratories. Intertek is a leading global testing, inspection, and certification laboratory. Funded by the US Grains Council 2 Primer: Vehicle Emissions of Toxic Compounds 3 Groups and Derivatives of Hydrocarbons Formaldehyde; Acetaldehyde Multiple Benzene Ethanol Rings = PAHs Benzene e.g. Benzopyrene Butadiene MTBE/ ETBE https://slideplayer.com/slide/4217696/ 4 Primer • Aldehyde: an organic compound containing the group —CHO, formed by the oxidation of alcohols. Typical aldehydes include methanal (formaldehyde) and ethanal (acetaldehyde). Many aldehydes are either gases or volatile liquids. • Aromatic Hydrocarbons: Aromatic hydrocarbons are those which contain one or more benzene rings. The name of the class comes from the fact that many of them have strong, pungent aromas. o Polycyclic aromatic hydrocarbons (PAHs, also polyaromatic hydrocarbons or polynuclear aromatic hydrocarbons: Are hydrocarbons—organic compounds containing only carbon and hydrogen—that are composed of multiple aromatic rings (organic rings in which the electrons are delocalized). -

Polycyclic Aromatic Hydrocarbons (Pahs) Polycyclic Aromatic Hydrocarbons (Pahs)
Polycyclic Aromatic Hydrocarbons (PAHs) Polycyclic Aromatic Hydrocarbons (PAHs) Polycyclic Aromatic Hydrocarbons (PAHs – also commonly called Polynuclear Aromatic Hydrocarbons) are compounds that are composed of multiple fused benzene rings. These compounds occur naturally in coal, crude oil, asphalt and gasoline. They are also formed by combustion of coal, oil, gas, wood, garbage and tobacco. PAHs have been identified as endocrine disruptors, and have been linked to several types of cancers. They are persistent in the environment, and can bioaccumulate. These compounds are regulated by many different agencies around the world to protect human health and the environment. Polycyclic Aromatic Hydrocarbons (PAHs) Compounds Compound CAS Matrix Cat. No. Unit Compound CAS. Matrix Cat. No. Unit Synonym Conc. Synonym Conc. Acenaphthene 83-32-9 NEAT H-108N 100 mg Dibenz[a,h]acridine 226-36-8 NEAT H-172N 10 mg 50 µg/mL Toluene H-108S 1 mL 50 µg/mL Toluene H-172S 1 mL Acenaphthylene 208-96-8 NEAT H-125N 100 mg Dibenz[a,j]acridine 224-42-0 NEAT H-173N 10 mg 50 µg/mL Toluene H-125S 1 mL 50 µg/mL Toluene H-173S 1 mL Acridine 260-94-6 NEAT H-187N 100 mg Dibenz[a,c]anthracene 215-58-7 NEAT H-134N 10 mg 50 µg/mL Toluene H-187S 1 mL 1,2:3,4-Dibenzanthracene 50 µg/mL Toluene H-134S 1 mL Anthanthrene 191-26-4 NEAT H-109N 10 mg Dibenz[a,h]anthracene 53-70-3 NEAT H-135N 10 mg 50 µg/mL Toluene H-109S 1 mL 1,2:5,6-Dibenzanthracene 50 µg/mL Toluene H-135S 1 mL Anthracene 120-12-7 NEAT H-110N 100 mg Dibenz[a,e]fluoranthene 5385-75-1 ---- ------ ----- 50 µg/mL Toluene -

Synonyms List
ENVIRONMENTAL CONTAMINANTS ENCYCLOPEDIA SYNONYMS LIST July 1, 1997 COMPILERS/EDITORS: ROY J. IRWIN, NATIONAL PARK SERVICE WITH ASSISTANCE FROM COLORADO STATE UNIVERSITY STUDENT ASSISTANT CONTAMINANTS SPECIALISTS: MARK VAN MOUWERIK LYNETTE STEVENS MARION DUBLER SEESE WENDY BASHAM NATIONAL PARK SERVICE WATER RESOURCES DIVISIONS, WATER OPERATIONS BRANCH 1201 Oakridge Drive, Suite 250 FORT COLLINS, COLORADO 80525 WARNING/DISCLAIMERS: Where specific products, books, or laboratories are mentioned, no official U.S. government endorsement is intended or implied. Digital format users: No software was independently developed for this project. Technical questions related to software should be directed to the manufacturer of whatever software is being used to read the files. Adobe Acrobat PDF files are supplied to allow use of this product with a wide variety of software, hardware, and operating systems (DOS, Windows, MAC, and UNIX). This document was put together by human beings, mostly by compiling or summarizing what other human beings have written. Therefore, it most likely contains some mistakes and/or potential misinterpretations and should be used primarily as a way to search quickly for basic information and information sources. It should not be viewed as an exhaustive, "last-word" source for critical applications (such as those requiring legally defensible information). For critical applications (such as litigation applications), it is best to use this document to find sources, and then to obtain the original documents and/or talk to the authors before depending too heavily on a particular piece of information. Like a library or many large databases (such as EPA's national STORET water quality database), this document contains information of variable quality from very diverse sources. -

Polycyclic Aromatic Hydrocarbons (Pahs) and to Emphasize the Human Health Effects That May Result from Exposure to Them
TOXICOLOGICAL PROFILE FOR POLYCYCLIC AROMATIC HYDROCARBONS U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry August 1995 PAHs ii DISCLAIMER The use of company or product name(s) is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry. PAHs iii UPDATE STATEMENT A Toxicological Profile for Polycyclic Aromatic Hydrocarbons was released in December 1990. This edition supersedes any previously released draft or final profile. Toxicological profiles are revised and republished as necessary, but no less than once every three years. For information regarding the update status of previously released profiles, contact ATSDR at: Agency for Toxic Substances and Disease Registry Division of Toxicology/Toxicology Information Branch 1600 Clifton Road NE, E-29 Atlanta, Georgia 30333 PAHs 1 1. PUBLIC HEALTH STATEMENT This statement was prepared to give you information about polycyclic aromatic hydrocarbons (PAHs) and to emphasize the human health effects that may result from exposure to them. The Environmental Protection Agency (EPA) has identified 1,408 hazardous waste sites as the most serious in the nation. These sites make up the National Priorities List (NPL) and are the sites targeted for long-term federal clean-up activities. PAHs have been found in at least 600 of the sites on the NPL. However, the number of NPL sites evaluated for PAHs is not known. As EPA evaluates more sites, the number of sites at which PAHs are found may increase. This information is important because exposure to PAHs may cause harmful health effects and because these sites are potential or actual sources of human exposure to PAHs.