The Correlated Electronic States of a Few Polycyclic Aromatic Hydrocarbons: a Computational Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

14 Title: Coal Review of Hydrocarbon Emissions Related to Tar Pitch
AP42 Section 7.1 Related: 14 Title: Review of Hydrocarbon Emissions Related to Coal Tar Pitch. Includes pitch vapor pressure data and Antoine's coefficients. EF Bart, et al. Allied Chemical Corporation 1980 I ._ ,.. ?i e!,/..'. r'.:.. ,,T.<,ili -:*., $0 ,i .# 1 .... i;.'.,.,: _..:, '">,U<>j. .. .. REVIEW OF HYDROCMSON EMISSIONS RELATED TO COAL TAR PITCH E. F. Bart, S. A. Visnic, P. A. Cerria Allied Chemical Corporation Morristown, New Jersey 07960 Sumnary Coal tar pitch is used primarily as a binder in the manufacture of carbon electrodes for the aluminum and steel industries. The pitch repre- sents the main product from the continuous, high-temperature distillation of coke oven tar. The pitch is produced and generally handled as a liquid at high temperatures, thus resulting in a potential for hydrocarbon emissions during its handling. An ever-increasing need for control of hydrocarbon emissions has resulted for process and storage equipment since amendments were made in 1977 to the Clean Air Act. Within the next several years, most industries will be faced with requirements for the installation of air pollution con- trol equipment. This paper deals with the types of emissions and methods used in quantifying and controlling emissions from coal tar pitch storage equipment. To better understand the nature of coal tar pitch, a brief description is given of its origin, chemistry and physical properties. Coal Tar Distillation Coal tar pitch is the residue from the distillation of coal tar and represents from 30% to 60% of the tar. It is a complex, bituminous sub- stance and has been estimated to contain about 5,000 compounds. -

C–H Arylation of Triphenylene, Naphthalene and Related Arenes Using Pd/C† Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue C–H arylation of triphenylene, naphthalene and related arenes using Pd/C† Cite this: Chem. Sci.,2015,6,1816 Karl D. Collins, Roman Honeker,‡ Suhelen Vasquez-C´ espedes,´ ‡ Dan-Tam D. Tang and Frank Glorius* A highly selective arylation of a number of polyaromatic hydrocarbons (PAHs) with aryliodonium salts and Pd/C as the only reagent is reported. The first C–H functionalization of triphenylene is explored, and proceeds at the most sterically hindered position. This non-chelate assisted C–H functionalization Received 4th October 2014 extends the reactivity profile of Pd/C and provides controlled access to p-extended PAHs, an important Accepted 19th December 2014 aspect of work towards the preparation of nanographenes. Mechanistic studies suggest in situ formation DOI: 10.1039/c4sc03051f of catalytically active insoluble nanoparticles, and that the reaction likely proceeds via a Pd(0)/Pd(II) type www.rsc.org/chemicalscience reaction manifold. Creative Commons Attribution 3.0 Unported Licence. Introduction precedent for the direct arylation of larger PAHs is reported. Seminal work by Oi and Inoue reported an effective arylation of Pd/C has long been established as an efficient catalyst in phenanthrene and uoranthene with aryltin trichlorides.10 One hydrogenation and cross-coupling reactions.1 Although sup- example of phenanthrene arylation has also been reported by ported catalysts are formally heterogeneous in nature, studies of Shi.11 Important studies by the group of Itami demonstrated a Pd/C suggest it typically acts as a reservoir of homogeneous more general solution (Scheme 1) using prepared boroxines as active catalytic species, following leaching of palladium from coupling partners.7d 1c,1e,2 the support into the solution. -

Polycyclic Aromatic Hydrocarbon Migration from Creosote-Treated Railway Ties Into Ballast and Adjacent Wetlands
United States In cooperation Department of with the Agriculture Polycyclic Aromatic United States Forest Service Department of Transportation Hydrocarbon Migration Forest Products Federal Laboratory Highway Administration From Creosote-Treated Research Paper FPL−RP−617 Railway Ties Into Ballast and Adjacent Wetlands Kenneth M. Brooks Abstract from the weathered ties at this time. No significant PAH loss was observed from ties during the second summer. A Occasionally, creosote-treated railroad ties need to be small portion of PAH appeared to move vertically down into replaced, sometimes in sensitive environments such as the ballast to approximately 60 cm. Small amounts of PAH wetlands. To help determine if this is detrimental to the may have migrated from the ballast into adjacent wetlands surrounding environment, more information is needed on during the second summer, but these amounts were not the extent and pattern of creosote, or more specifically poly- statistically significant. These results suggest that it is rea- cyclic aromatic hydrocarbon (PAH), migration from railroad sonable to expect a detectable migration of creosote-derived ties and what effects this would have on the surrounding PAH from newly treated railway ties into supporting ballast environment. This study is a report on PAH level testing during their first exposure to hot summer weather. The PAH done in a simulated wetland mesocosm. Both newly treated rapidly disappeared from the ballast during the fall and and weathered creosote-treated railroad ties were placed in winter following this initial loss. Then statistically insignifi- the simulated wetland. As a control, untreated ties were also cant vertical and horizontal migration of these PAH suggests placed in the mesocosm. -

Safeguards Benzopyrene Contamination in Korean
SAFEGUARDS SGS CONSUMER TESTING SERVICES FOOD NO. 187/12 NOV 2012 BENZOPYRENE CONTAMINATION IN KOREAN INSTANT NOODLE On 25 Oct 2012, Korea Food and Drug Administration (KFDA) recalled six instant noodle products from the nation’s largest ramen maker in South Korea because these products were found to be tainted with benzo(a) pyrene, which is a known carcinogen.1 Several countries, including Philippines, Thailand, Taiwan and China, have been alerted to this and as a precaution, have removed these products from their shelves. Benzo(a)pyrene belongs to the group of polycyclic aromatic hydrocarbons (PAHs). Most PAHs derive from incomplete burning of carbon containing materials such as petroleum fuels, wood, and garbage. The occurrence of PAHs in foods is mainly caused by environmental contamination (water, air and soil), food processing (direct drying and heating), and packaging materials. PAHs have been found in different food products, such as dairy products, vegetables, fruits, oil, coffee, tea, cereals, smoked meat, seafood products, ingredients for food formulation, seasoning, and instant noodles. announcement of the test results and the recall order because the levels of In June, the KFDA conducted tests on benzopyrene found in the noodle products in June were minuscule and not harmful, 30 instant noodle products sold in the South Korean agency reported. However, the KFDA issued the recall order after South Korea and found that six its commissioner Lee Hee-sung was pressed on the matter during a legislative products contained benzopyrene, with question-and-answer, the report indicated.2 one product containing the highest level of 4.7 parts per billion (µg/kg). -

Polycyclic Aromatic Hydrocarbon Structure Index
NIST Special Publication 922 Polycyclic Aromatic Hydrocarbon Structure Index Lane C. Sander and Stephen A. Wise Chemical Science and Technology Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899-0001 December 1997 revised August 2020 U.S. Department of Commerce William M. Daley, Secretary Technology Administration Gary R. Bachula, Acting Under Secretary for Technology National Institute of Standards and Technology Raymond G. Kammer, Director Polycyclic Aromatic Hydrocarbon Structure Index Lane C. Sander and Stephen A. Wise Chemical Science and Technology Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899 This tabulation is presented as an aid in the identification of the chemical structures of polycyclic aromatic hydrocarbons (PAHs). The Structure Index consists of two parts: (1) a cross index of named PAHs listed in alphabetical order, and (2) chemical structures including ring numbering, name(s), Chemical Abstract Service (CAS) Registry numbers, chemical formulas, molecular weights, and length-to-breadth ratios (L/B) and shape descriptors of PAHs listed in order of increasing molecular weight. Where possible, synonyms (including those employing alternate and/or obsolete naming conventions) have been included. Synonyms used in the Structure Index were compiled from a variety of sources including “Polynuclear Aromatic Hydrocarbons Nomenclature Guide,” by Loening, et al. [1], “Analytical Chemistry of Polycyclic Aromatic Compounds,” by Lee et al. [2], “Calculated Molecular Properties of Polycyclic Aromatic Hydrocarbons,” by Hites and Simonsick [3], “Handbook of Polycyclic Hydrocarbons,” by J. R. Dias [4], “The Ring Index,” by Patterson and Capell [5], “CAS 12th Collective Index,” [6] and “Aldrich Structure Index” [7]. In this publication the IUPAC preferred name is shown in large or bold type. -

The Study of the Absoiption Spectra of the Hydrocarbons Isolated from the Products of the Action of Alunzinium Chloride on Nup12tkccle.R~E
View Article Online / Journal Homepage / Table of Contents for this issue ABSORPTION SPECTRA OF HYDROCARBONS. 1319 Published on 01 January 1908. Downloaded by University of California - San Diego 12/04/2016 06:21:24. CXXV1.-The Study of the Absoiption Spectra of the Hydrocarbons isolated from the Products of the Action of Alunzinium Chloride on Nup12tkccle.r~e. By ANNIEHOMER, Fellow of Newnham College, and JOHNEDWARD PURVIS, M.A. FROMthe products of the action of aluminium chloride on naphthalene, besides PP-dinaphthyl previously isolated by Friedel and Crafts from the same reaction, there have been isolated three new hydrocarbons which have been described in detail by one of us (Homer, Trans., 1907, 9 1, 1103). The substances isolated were : (.i) CI4Hl6, a homologue of naphthalene, either tetramethyl- or 4s2 View Article Online 1320 HOMER AND PURVIS: THE STUDY OF THE diethyl-naphthalene, more probably the former ; (ii) CZ0Hl4, /3@-dinaphthyl; (iii) c26H22, a substance supposed to be a homologue of dinaphthanthracene, C,,H14 ; and (iv) C40H26, probably tetra- naph t h yl. @B-Dinaphthylis formed by the condensation of two naphthalene molecules. It was thought that the hydrocarbon C,,HZ6 was formed by the condensation of either two PP-dinaphthgl or four naphthalene molecules, more probably by the former, as an increase in the time allowed for the action of aluminium chloride on naphthalene, or a rise in the temperature at which the reaction was conducted, caused a decrease in the amount of the hydrocarbon aad an increase in the amount of the hydrocarbon C,,H,, produced. It was suggested that the substance C261322was a homologue of dinaphthanthracene, CZ2Hl4,and its formation from alkylnaphthalenes, which are also formed during the reaction, was given as follows : The intense fluorescence of the substance suggested the presence of an anthracenoid linking, In the method of formation thus proposed, hydrogen would be eliminated from a /3-methyl group of trimethylnaphthalene. -

Determination of Petroleum Hydrocarbons in Sediments
UNITED NATIONS ENVIRONMENT PROGRAMME NOVEMBER 1992 Determination of petroleum hydrocarbons in sediments Reference Methods For Marine Pollution Studies No. 20 Prepared in co-operation with IOC IAEA UNEP 1992 ~ i - PREFACE The Regional Seas Programme was initiated by UNEP in 1974. Since then the Governing Conncil ofUNEP has repeatedly endorsed a regional approach to the control of marine pollution and the management of marine and coastal resources and has requested the development of regional action plans. The Regional Seas Progranune at present includes ten regions and has over 120 coastal States participating in it (1),(2). One of the basic components of the action plans sponsored by UNEP in the framework of the Regional Seas Programme is the assessment of the state of the marine em~ronment and of its resources, and of the sources and trends of the pollution, and the impact of pollution on human health, marine ecosystems and amenities. In order to assist those participating in this activity and to ensure that the data obtained through this assessment can be compared. on a world-wide basis and thns contribute to the Global Environment Monitoring System (GEMS) of UNEP, a set of Reference Methods and Guidelines for marine pollution studies is being developed as part of a programme of c9mprehensive technical support which includes the provision of expert advice, reference methods and materials, training and data quality assurance (3). The Methods are recommended to be adopted by Governments participating in tbe Regional Seas Programme. The methods and guidelines are prepared in co-operation with the relevant specialized bodies of the United Nations system as well as other organizations and are tested by a number of experts competent in the field relevant to the methods described. -

Analysis of Benzopyrenes and Benzopyrene Metabolites by Fluorescence Spectroscopy Techniques
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2016 Analysis of Benzopyrenes and Benzopyrene Metabolites by Fluorescence Spectroscopy Techniques Bassam Al-Farhani University of Central Florida Part of the Chemistry Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Doctoral Dissertation (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Al-Farhani, Bassam, "Analysis of Benzopyrenes and Benzopyrene Metabolites by Fluorescence Spectroscopy Techniques" (2016). Electronic Theses and Dissertations, 2004-2019. 5286. https://stars.library.ucf.edu/etd/5286 ANALYSIS OF BENZOPYRENES AND BENZOPYRENE METABOLITES BY FLUORESCENCE SPECTROSCOPY TECHNIQUES by BASSAM AL-FARHANI B.Sc. Alnahrain University Iraq, 2002 M.Sc. Alnahrain University Iraq, 2005 A dissertation submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry in the College of Sciences at the University of Central Florida Orlando, Florida Spring Term 2016 Major Professor: Andres D. Campiglia © 2016 Bassam Al-farhani ii ABSTRACT Polycyclic aromatic hydrocarbons (PAHs) are some of the most common and toxic pollutants encountered worldwide. Presently, monitoring is restricted to sixteen PAHs, but it is well understood that this list omits many toxic PAHs. Among the “forgotten” PAHs, isomers with molecular weight 302 are of particular concern due to their high toxicological properties. The chromatographic analysis of PAHs with MW 302 is challenged by similar retention times and virtually identical mass fragmentation patterns. -

Ambient Water Quality Criteria for Polycyclic Aromatic Hydrocarbons (Pahs)
Ambient Water Quality Criteria For Polycyclic Aromatic Hydrocarbons (PAHs) Ministry of Environment, Lands and Parks Province of British Columbia N. K. Nagpal, Ph.D. Water Quality Branch Water Management Division February, 1993 ACKNOWLEDGEMENTS The author is indebted to the following individual and agencies for providing valuable comments during the preparation of this document. Dr. Ray Copes BC. Ministry of Health, Victoria, BC. Dr. G. R. Fox Environmental Protection Div., BC. MOELP, Victoria, BC. Mr. L. W. Pommen Water Quality Branch, BC. MOELP, Victoria, BC. Mr. R. J. Rocchini Water Quality Branch, BC. MOELP, Victoria, BC. Ms. Sherry Smith Eco-Health Branch, Conservation and Protection, Environment Canada, Hull, Quebec Mr. Scott Teed Eco-Health Branch, Conservation and Protection, Environment Canada, Hull, Quebec Ms. Bev Raymond Integrated Programs Branch, Inland Waters, Environment Canada, North Vancouver, BC. 1.0 INTRODUCTION Polycyclic aromatic hydrocarbons (PAHs) are organic compounds which are non- essential for the growth of plants, animals or humans; yet, they are ubiquitous in the environment. When present in sufficient quantity in the environment, certain PAHs are toxic and carcinogenic to plants, animals and humans. This document discusses the characteristics of PAHs and their effects on various water uses, which include drinking Ministry of Environment Water Protection and Sustainability Branch Mailing Address: Telephone: 250 387-9481 Environmental Sustainability PO Box 9362 Facsimile: 250 356-1202 and Strategic Policy Division Stn Prov Govt Website: www.gov.bc.ca/water Victoria BC V8W 9M2 water, aquatic life, wildlife, livestock watering, irrigation, recreation and aesthetics, and industrial water supplies. A significant portion of this document discusses the effects of PAHs upon aquatic life, due to its sensitivity to PAHs. -

Absence of Photoemission from the Fermi Level in Potassium Intercalated Picene and Coronene films: Structure, Polaron Or Correlation Physics?
Absence of photoemission from the Fermi level in potassium intercalated picene and coronene films: structure, polaron or correlation physics? Benjamin Mahns,1 Friedrich Roth,1 and Martin Knupfer1 IFW Dresden, P.O. Box 270116, D-01171 Dresden, Germany (Dated: 13 September 2018) The electronic structure of potassium intercalated picene and coronene films has been studied using photoemission spectroscopy. Picene has additionally been intercalated using sodium. Upon alkali metal addition core level as well as valence band pho- toemission data signal a filling of previously unoccupied states of the two molecular materials due to charge transfer from potassium. In contrast to the observation of superconductivity in Kxpicene and Kxcoronene (x ∼ 3), none of the films studied shows emission from the Fermi level, i. e. we find no indication for a metallic ground state. Several reasons for this observation are discussed. arXiv:1203.4728v1 [cond-mat.supr-con] 21 Mar 2012 1 I. INTRODUCTION Superconductivity always has attracted a large number of researchers since this phe- nomenon harbors challenges and prospects both under fundamental and applied points of view. Very recently, it has been discovered that some molecular crystal consisting of poly- cyclic aromatic hydrocarbons demonstrate superconductivity upon alkali metal addition. Furthermore, these compounds are characterized by rather high transition temperatures into 1 2 the superconducting state, for instance K3picene (Tc=18 K) and K3coronene (Tc=15 K) . Most recently, superconductivity with a transition temperature of 33 K has been reported 3 for K3dibenzopentacene . These doped aromatic hydrocarbons thus represent a class of or- ganic superconductors with transition temperatures only slightly below that of the famous 4{8 alkali metal doped fullerenes with Tc's up to 40 K . -

Laboratory Spectroscopy and Astronomical Significance of The
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Repository@Nottingham Laboratory spectroscopy and astronomical significance of the fully-benzenoid PAH triphenylene and its cation V. Kofmana,b, P.J. Sarrec, R.E. Hibbinsc,d, I.L. ten Kateb, H. Linnartza aSackler Laboratory for Astrophysics, Leiden Observatory, Leiden University, PO Box 9513, 2300 RA Leiden, The Netherlands bDepartment of Earth Sciences, Utrecht University, Budapestlaan 4, 3584 CD Utrecht, The Netherlands cSchool of Chemistry, The University of Nottingham, University Park, Nottingham NG7 2RD, United Kingdom dDepartment of Physics, Norwegian University of Science and Technology, N-7491 Trondheim, Norway Abstract Triphenylene (C18H12) is a highly symmetric polycyclic aromatic hydrocarbon (PAH) molecule with a `fully-benzenoid' electronic structure. This confers a high chemical stability compared with PAHs of similar size. Although numerous infrared and UV-visible experimental spectroscopic and theoretical studies of a wide range PAHs in an astrophysical context have been conducted, triphenylene and its radical cation have received almost no attention. There exists a huge body of spectroscopic evidence for neutral and ionised PAHs in astrophysical sources, obtained principally through detection of infrared emission features that are characteristic of PAHs as a chemical class. However, it has so far not proved possible to identify spectroscopically a single isolated PAH in space, although PAHs including triphenylene have been detected -
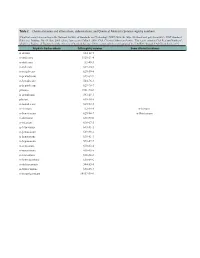
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No.