Metanephrops Challengeri)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lobsters-Identification, World Distribution, and U.S. Trade
Lobsters-Identification, World Distribution, and U.S. Trade AUSTIN B. WILLIAMS Introduction tons to pounds to conform with US. tinents and islands, shoal platforms, and fishery statistics). This total includes certain seamounts (Fig. 1 and 2). More Lobsters are valued throughout the clawed lobsters, spiny and flat lobsters, over, the world distribution of these world as prime seafood items wherever and squat lobsters or langostinos (Tables animals can also be divided rougWy into they are caught, sold, or consumed. 1 and 2). temperate, subtropical, and tropical Basically, three kinds are marketed for Fisheries for these animals are de temperature zones. From such partition food, the clawed lobsters (superfamily cidedly concentrated in certain areas of ing, the following facts regarding lob Nephropoidea), the squat lobsters the world because of species distribu ster fisheries emerge. (family Galatheidae), and the spiny or tion, and this can be recognized by Clawed lobster fisheries (superfamily nonclawed lobsters (superfamily noting regional and species catches. The Nephropoidea) are concentrated in the Palinuroidea) . Food and Agriculture Organization of temperate North Atlantic region, al The US. market in clawed lobsters is the United Nations (FAO) has divided though there is minor fishing for them dominated by whole living American the world into 27 major fishing areas for in cooler waters at the edge of the con lobsters, Homarus americanus, caught the purpose of reporting fishery statis tinental platform in the Gul f of Mexico, off the northeastern United States and tics. Nineteen of these are marine fish Caribbean Sea (Roe, 1966), western southeastern Canada, but certain ing areas, but lobster distribution is South Atlantic along the coast of Brazil, smaller species of clawed lobsters from restricted to only 14 of them, i.e. -

29 November 2005
University of Auckland Institute of Marine Science Publications List maintained by Richard Taylor. Last updated: 31 July 2019. This map shows the relative frequencies of words in the publication titles listed below (1966-Nov. 2017), with “New Zealand” removed (otherwise it dominates), and variants of stem words and taxonomic synonyms amalgamated (e.g., ecology/ecological, Chrysophrys/Pagrus). It was created using Jonathan Feinberg’s utility at www.wordle.net. In press Markic, A., Gaertner, J.-C., Gaertner-Mazouni, N., Koelmans, A.A. Plastic ingestion by marine fish in the wild. Critical Reviews in Environmental Science and Technology. McArley, T.J., Hickey, A.J.R., Wallace, L., Kunzmann, A., Herbert, N.A. Intertidal triplefin fishes have a lower critical oxygen tension (Pcrit), higher maximal aerobic capacity, and higher tissue glycogen stores than their subtidal counterparts. Journal of Comparative Physiology B: Biochemical, Systemic, and Environmental Physiology. O'Rorke, R., Lavery, S.D., Wang, M., Gallego, R., Waite, A.M., Beckley, L.E., Thompson, P.A., Jeffs, A.G. Phyllosomata associated with large gelatinous zooplankton: hitching rides and stealing bites. ICES Journal of Marine Science. Sayre, R., Noble, S., Hamann, S., Smith, R., Wright, D., Breyer, S., Butler, K., Van Graafeiland, K., Frye, C., Karagulle, D., Hopkins, D., Stephens, D., Kelly, K., Basher, Z., Burton, D., Cress, J., Atkins, K., Van Sistine, D.P., Friesen, B., Allee, R., Allen, T., Aniello, P., Asaad, I., Costello, M.J., Goodin, K., Harris, P., Kavanaugh, M., Lillis, H., Manca, E., Muller-Karger, F., Nyberg, B., Parsons, R., Saarinen, J., Steiner, J., Reed, A. A new 30 meter resolution global shoreline vector and associated global islands database for the development of standardized ecological coastal units. -

A Synthesis of Existing Data on Larval Rock Lobster Distribution in Southern Australia Project 96/107
A synthesis of existing data on larval rock lobster distribution in southern Australia Project 96/107 DEVELOPMENT CORPORATION Final Report to FRDC CS I RO Final Report the Project 96/107 B. Bruce, R. Bradford, 0. Griffin, C. Gardner and J. Young CSIRO Marine Laboratories Hobart ISBN: 0 643 062270 FRDC FIN.AL REPORT: PROJECT NO. 96/107 3, 5. 5.1 Scope and Rationale of 5.2 Sources of data {collaborating institutions) ......................................................................... 10 5.3 Sample Coverage ............................................................................................................... i2 5.3.1 Geographic ................................................................................................................................................. 12 5.3.2 Annual ........................................................................................................................................................ 13 5.3.3 Seasonal .................................................................................................................................................... 13 5.4 Net Systems: ...................................................................................................................... 14 5.4.1 Surface sampling ........................................................................................................................................ 14 Surface net ..................................................................................................................................... -

(Jasus Edwardsii Hutton, 1875) Larvae
Environmental Physiology of Cultured Early-Stage Southern Rock Lobster (Jasus edwardsii Hutton, 1875) Larvae Michel Francois Marie Bermudes Submitted in fulfilment of the requirements for the degree of Doctor Of Philosophy University of Tasmania November 2002 Declarations This thesis contains no material which has been accepted for a degree or diploma by the University or any other institution, except by way of background information in duly acknowledged in the thesis, and to the best of the candidate's knowledge and belief, no material previously published or written by another person except where due acknowledgment is made in the text of the thesis. Michel Francois Marie Bermudes This thesis may be available for loan and limited copying in accordance with the Copyright Act 1968. Michel Francois Marie Bermudes Abstract The aim of this project was to define more clearly the culture conditions for the propagation of the southern rock lobster (Jasus echvardsii) in relation to environmental bioenergetic constraints. The effects of temperature and photoperiod on the first three stages of development were first studied in small-scale culture experiments. Larvae reared at 18°C developed faster and reached a larger size at stage IV than larvae cultured at 14°C. Development through stage II was shorter under continuous light. However, the pattern of response to photoperiod shifted at stage III when growth was highest in all the light/dark phase treatments than under continuous light. The influence of temperature and light intensity in early-stage larvae was further investigated through behavioural and physiological studies. Results obtained in stages I, II and III larvae indicated an energetic imbalance at high temperature (-22°C). -

Large Spiny Lobsters Reduce the Catchability of Small Conspecifics
Vol. 666: 99–113, 2021 MARINE ECOLOGY PROGRESS SERIES Published May 20 https://doi.org/10.3354/meps13695 Mar Ecol Prog Ser OPEN ACCESS Size matters: large spiny lobsters reduce the catchability of small conspecifics Emma-Jade Tuffley1,2,3,*, Simon de Lestang2, Jason How2, Tim Langlois1,3 1School of Biological Sciences, The University of Western Australia, 35 Stirling Highway, Crawley, WA 6009, Australia 2Aquatic Science and Assessment, Department of Primary Industries and Regional Development, 39 Northside Drive, Hillarys, WA 6025, Australia 3The UWA Oceans Institute, Indian Ocean Marine Research Centre, Cnr. of Fairway and Service Road 4, Crawley, WA 6009, Australia ABSTRACT: Indices of lobster abundance and population demography are often derived from pot catch rate data and rely upon constant catchability. However, there is evidence in clawed lobsters, and some spiny lobsters, that catchability is affected by conspecifics present in pots, and that this effect is sex- and size-dependent. For the first time, this study investigated this effect in Panulirus cyg nus, an economically important spiny lobster species endemic to Western Australia. Three studies: (1) aquaria trials, (2) pot seeding experiments, and (3) field surveys, were used to investi- gate how the presence of large male and female conspecifics influence catchability in smaller, immature P. cygnus. Large P. cygnus generally reduced the catchability of small conspecifics; large males by 26−33% and large females by 14−27%. The effect of large females was complex and varied seasonally, dependent on the sex of the small lobster. Conspecific-related catchability should be a vital consideration when interpreting the results of pot-based surveys, especially if population demo graphy changes. -

The Fossil Clawed Lobster, Metanephrops Elongatus Hu & Tao
Zootaxa 3760 (3): 494–496 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Correspondence ZOOTAXA Copyright © 2014 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3760.3.17 http://zoobank.org/urn:lsid:zoobank.org:pub:0445CBDC-C2A9-466A-9A0B-FBE706E2A5DC Taxonomic note: the fossil clawed lobster, Metanephrops elongatus Hu & Tao, 2000 (Nephropidae), is a nomen dubium but definitely not a Metanephrops DALE TSHUDY1 & TIN-YAM CHAN2 1Department of Geosciences, Edinboro University of Pennsylvania, Edinboro, Pennsylvania 16412, U.S.A. E-mail: [email protected] 2Institute of Marine Biology and Center of Excellence for the Oceans, National Taiwan Ocean University, Keelung 202, Taiwan. E-mail: [email protected] Metanephrops is an extant, clawed lobster genus (Family Nephropidae) with a very distinctive, carinate and spiny cephalothorax. It is the most speciose extant genus of clawed lobster, with 18 Recent species known. It is a deepwater genus with very large eyes; most species are known from deeper than 150 meters. Some species, nonetheless, are of commercial importance and marketed as “scampi”. Here, we report that the fossil species, Metanephrops elongatus Hu and Tao, 2000, does not belong to Metanephrops and is a nomen dubium. Metanephrops elongatus was erected based on a single, fragmentary fossil specimen from Pliocene rocks of Central Taiwan. Its validity has not been questioned previously in the literature and, thus, it has been included in recent compilations of lobster species (e.g. De Grave et al. 2009; Schweitzer et al. 2010). This sole specimen and holotype is deposited in the private collections of the Land Fossils and Minerals Museum, Tainan, Taiwan, where we examined and photographed it in May, 2013. -

Effect of Starvation on the First Larvae of <I>Homarus Americanus</I>
BULLETIN OF MARINE SCIENCE, 61(1): 73–80, 1997 EFFECT OF STARVATION ON THE FIRST LARVAE OF HOMARUS AMERICANUS (DECAPODA, NEPHROPIDAE) AND PHYLLOSOMAS OF JASUS VERREAUXI AND J. EDWARDSII (DECAPODA, PALINURIDAE) Fernando A. Abrunhosa and Jiro Kittaka ABSTRACT Food is one of the important factors controlling decapod larval culture, however, little is known about the effect of the starvation regimen on the physiological condition of the lar- vae. In the present study, the influence of starvation upon survival rate and the intermolt period was observed in the first instar of the American lobster, Homarus americanus, the first instars of phyllosomas of the red rock lobster, Jasus edwardsii, and the green rock lobster, J. verreauxi. Larvae were reared in receptacles of 150 ml capacity filled with sea water and submitted to two feeding regimens: larvae were submitted to an initial period of starvation and larvae were submitted to an initial period of feeding. Larvae of H. americanus were cultured individually at 17 - 18°C, while phyllosomas were cultured at five larvae per receptacle at temperatures of 19 - 22°C and 16 - 17° C for J. edwardsii and J. verreauxi, respectively. No larvae succeeded in molting if completely starved or if they were fed after a prolonged starvation period. However, the species showed a period of tolerance before food was introduced. The average interval between the first day of feeding and the first day of molting was relatively constant within each species: about 4, 10 and 12 d for H. americanus, J. edwardsii and J. verreauxi, respectively. The starvation tolerance period (50% survival) averaged about 5, 4 and 8 d for these species, respectively. -

Annotated Checklist of New Zealand Decapoda (Arthropoda: Crustacea)
Tuhinga 22: 171–272 Copyright © Museum of New Zealand Te Papa Tongarewa (2011) Annotated checklist of New Zealand Decapoda (Arthropoda: Crustacea) John C. Yaldwyn† and W. Richard Webber* † Research Associate, Museum of New Zealand Te Papa Tongarewa. Deceased October 2005 * Museum of New Zealand Te Papa Tongarewa, PO Box 467, Wellington, New Zealand ([email protected]) (Manuscript completed for publication by second author) ABSTRACT: A checklist of the Recent Decapoda (shrimps, prawns, lobsters, crayfish and crabs) of the New Zealand region is given. It includes 488 named species in 90 families, with 153 (31%) of the species considered endemic. References to New Zealand records and other significant references are given for all species previously recorded from New Zealand. The location of New Zealand material is given for a number of species first recorded in the New Zealand Inventory of Biodiversity but with no further data. Information on geographical distribution, habitat range and, in some cases, depth range and colour are given for each species. KEYWORDS: Decapoda, New Zealand, checklist, annotated checklist, shrimp, prawn, lobster, crab. Contents Introduction Methods Checklist of New Zealand Decapoda Suborder DENDROBRANCHIATA Bate, 1888 ..................................... 178 Superfamily PENAEOIDEA Rafinesque, 1815.............................. 178 Family ARISTEIDAE Wood-Mason & Alcock, 1891..................... 178 Family BENTHESICYMIDAE Wood-Mason & Alcock, 1891 .......... 180 Family PENAEIDAE Rafinesque, 1815 .................................. -

Developmental Changes in the Mouthparts of Juvenile Caribbean Spiny Lobster, Panulirus Argus: Implications for Aquaculture
FAD LIBRARIES Florida Atlantic University HARBOR BRANCH ~ FLORIDA ATLANTIC UNIVERSITY FAU Institutional Repository http://purl.fcla.edu/fau/fauir This paper was submitted by the faculty of FAU’s Harbor Branch Oceanographic Institute. Notice: ©2008 Elsevier B.V. The final published version of this manuscript is available at http://www.sciencedirect.com/science/journal/00448486. This article may be cited as: Cox, S. L., Jeffs, A. G., & Davis, M. (2008). Developmental changes in the mouthparts of juvenile Caribbean spiny lobster, Panulirus argus: Implications for aquaculture. Aquaculture, 283(1‐4), 168‐174. doi:10.1016/j.aquaculture.2008.07.019 Aquaculture 283 (2008) 168–174 Contents lists available at ScienceDirect Aquaculture journal homepage: www.elsevier.com/locate/aqua-online Developmental changes in the mouthparts of juvenile Caribbean spiny lobster, Panulirus argus: Implications for aquaculture Serena L. Cox a,b,⁎, Andrew G. Jeffs c, Megan Davis a a Aquaculture Division, Harbor Branch Oceanographic Institute at Florida Atlantic University, 5600 US 1 North, Fort Pierce, Florida 34946, USA b National Institute of Water and Atmospheric Research, Private Bag 14901, Kilbirnie, Wellington 6241, New Zealand c Leigh Marine Laboratory, University of Auckland, P.O. Box 349, Warkworth 0941, New Zealand article info abstract Article history: Light microscopy and video analysis were used to examine the mouthpart morphology and feeding Received 5 May 2008 behaviour of the Caribbean spiny lobster from puerulus (megalopal stage) (5–8 mm carapace length, CL) to Received in revised form 14 July 2008 adult (85 mm CL). Upon settlement the pueruli did not possess fully functional mouthparts, however, Accepted 14 July 2008 efficient feeding appendages appeared in the first instar juvenile (after the first moult from puerulus). -
![[I]Metanephrops Challengeri[I]](https://docslib.b-cdn.net/cover/8281/i-metanephrops-challengeri-i-1598281.webp)
[I]Metanephrops Challengeri[I]
A peer-reviewed version of this preprint was published in PeerJ on 20 September 2018. View the peer-reviewed version (peerj.com/articles/5641), which is the preferred citable publication unless you specifically need to cite this preprint. van der Reis AL, Laroche O, Jeffs AG, Lavery SD. 2018. Preliminary analysis of New Zealand scampi (Metanephrops challengeri) diet using metabarcoding. PeerJ 6:e5641 https://doi.org/10.7717/peerj.5641 Preliminary analysis of New Zealand scampi (Metanephrops challengeri) diet using metabarcoding Aimee L van der Reis Corresp., 1 , Olivier Laroche 2 , Andrew G Jeffs 1, 2 , Shane D Lavery 1, 2 1 Institute of Marine Science, University of Auckland, Auckland, New Zealand 2 School of Biological Sciences, University of Auckland, Auckland, New Zealand Corresponding Author: Aimee L van der Reis Email address: [email protected] Deep sea lobsters are highly valued for seafood and provide the basis of important commercial fisheries in many parts of the world. Despite their economic significance, relatively little is known about their natural diets. Microscopic analyses of foregut content in some species have suffered from low taxonomic resolution, with many of the dietary items difficult to reliably identify as their tissue is easily digested. DNA metabarcoding has the potential to provide greater taxonomic resolution of the diet of the New Zealand scampi (Metanephrops challengeri) through the identification of gut contents, but a number of methodological concerns need to be overcome first to ensure optimum DNA metabarcoding results. In this study, a range of methodological parameters were tested to determine the optimum protocols for DNA metabarcoding, and provide a first view of M. -

Associate Professor Caleb Gardner
Curriculum Vitae A ssoci ate Prof essor Caleb Gardner Contents 1. Summary 2 2. Personal Details 2 3. Qualifications 2 4. Current Employment 3 5. External Grants 3 6. Current Committee Membership 7 7. Refereed Publications 8 8. Research and Management Reports 14 9. Students 20 Caleb Gardner 16/12/2013 Page 1 Summary I have qualifications in both economics and biology which interact in research on commercial fisheries. I currently hold two positions. My main role is as the Director, Sustainable Marine Research Collaboration Agreement (SMRCA), Institute for Marine and Antarctic Studies, UTAS in South East Australia. This role involves supervision and resourcing of over 60 staff and 38 PhD students operating across around 150 projects. Research is mainly on the larger marine industries of farmed Atlantic salmon and wild harvest blacklip abalone and southern rock lobster. However activities also span many other operations including recreational fisheries, scalefish, crabs, scallops and oyster culture. In addition to my role as Director SMRCA, I lead several research projects dealing with wild fisheries species, generally with the objective of improving harvest strategies. I also have a smaller role in leading research activities on wild harvest fisheries at the Australian Seafood Cooperative Research Centre. Projects in this organisation are mainly related to improving economic yield and reducing ecosystem impacts through better management. Projects involve partnerships between research organisations around Australia and industry organisations including western rock lobster, southern rock lobster, abalone, finfish and prawn fisheries. Personal Details Name: Associate Professor Caleb Gardner Address: 2 Jersey St, Sandy Bay, 7005 Phone: H- +61 (03) 6224 8417 W- +61 (03) 6227 7233 Mob- 0409 427 366 Fax- +61 (03) 6227 8035 Email: [email protected] Qualifications • Bachelor of Science. -
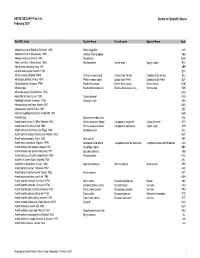
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste