Sustainable Synthesis and Polycondensation Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dehydration of Cellulose to Levoglucosenone Using Polar Aprotic Solvents Cite This: Energy Environ
Energy & Environmental Science View Article Online PAPER View Journal | View Issue Dehydration of cellulose to levoglucosenone using polar aprotic solvents Cite this: Energy Environ. Sci., 2015, 8,1808 Fei Cao,ab Thomas J. Schwartz,a Daniel J. McClelland,a Siddarth H. Krishna,a James A. Dumesica and George W. Huber*a Herein, we report an approach to produce levoglucosenone (LGO) from cellulose in yields up to 51% under Received 2nd February 2015, mild reaction conditions (170–230 1C; 5–20 mM H2SO4) using polar, aprotic solvents such as tetrahydro- Accepted 18th May 2015 furan(THF).LGOcanbeusedtomakeawidevarietyof chemicals from biomass. The water content and DOI: 10.1039/c5ee00353a solvent used in the reaction system control the product distribution. LGO is produced from the dehydration of levoglucosan (LGA). LGA is produced from cellulose depolymerization. Increasing the water content leads www.rsc.org/ees to the production of 5-hydroxymethyl furfural (HMF), obtaining a maximum HMF yield of 30%. Creative Commons Attribution 3.0 Unported Licence. Broader context This article describes the sustainable conversion of cellulose to levoglucosenone (LGO) using mild reaction conditions in the liquid phase. LGO is an attractive, biomass-derived platform molecule that can be used for the renewable production of pharmaceuticals, commodity chemicals such as 1,6-hexanediol (HDO), and green solvents such as dihydrolevoglucosenone. The co-production of such high-value species has the potential to significantly improve the overall economic feasibility of a biorefinery. Notably, dihydrolevoglucosenone, also known as Cyrenes, is a polar, aprotic solvent with properties similar to N- methylpyrrolidone and sulfolane, both of which are traditionally obtained from fossil-based resources. -

Aldrichimica Acta
VOLUME 51, NO. 1 | 2018 ALDRICHIMICA ACTA The Spectacular Resurgence of Electrochemical Redox Reactions in Organic Synthesis Carbon–Carbon π Bonds as Conjunctive Reagents in Cross-Coupling The life science business of Merck KGaA, Darmstadt, Germany operates as MilliporeSigma in the U.S. and Canada. MakInG sTrIDes In Genome EDITInG: THe DIGITaL, CHemIcaL, anD BIoLogIcaL Dear Fellow Researchers: You may not know that MilliporeSigma can make DNA from scratch, using 100% synthetic chemical methods—no living cells and no fermentation necessary. Our DNA synthesis facilities in The Woodlands (Texas) and Haverhill (U.K.) receive thousands of DNA orders daily from scientists all over the world. A small piece of DNA, one hundred bases long, gives scientists 4100 possibilities using only the fundamental letters of our genetic code (A,C,G,T). Included within these options is the opportunity for a researcher to open a common text editor on his or her PC and design a 100-base piece of DNA. This piece of DNA could help study diseases ranging from sickle cell anemia and blindness to cystic fibrosis by using CRISPR. To get started, scientists cut this 100-base DNA text from the text editor and paste it into MilliporeSigma’s online ordering system. The corresponding DNA piece that likely has never existed before is then delivered to them in 24–48 hours. Once received, this piece of DNA is mixed with CRISPR and living cells, and, with a “blast” of electricity, these synthetic chemical pieces activate the cell to edit the genome. Learn more about our CRISPR gene editing portfolio at SigmaAldrich.com/CRISPR Sincerely yours, Udit Batra, Ph.D. -

The Decomposition Kinetics of Peracetic Acid and Hydrogen Peroxide in Municipal Wastewaters
Disinfection Forum No 10, October 2015 The Decomposition Kinetics of Peracetic Acid and Hydrogen Peroxide in Municipal Wastewaters INTRODUCTION Efficient control of microbial populations in municipal wastewater using peracetic acid (PAA) requires an understanding of the PAA decomposition kinetics. This knowledge is critical to ensure the proper dosing of PAA needed to achieve an adequate concentration within the contact time of the disinfection chamber. In addition, the impact of PAA on the environment, post-discharge into the receiving water body, also is dependent upon the longevity of the PAA in the environment, before decomposing to acetic acid, oxygen and water. As a result, the decomposition kinetics of PAA may have a significant impact on aquatic and environmental toxicity. PAA is not manufactured as a pure compound. The solution exists as an equilibrium mixture of PAA, hydrogen peroxide, acetic acid, and water: ↔ + + Acetic Acid Hydrogen Peroxide Peracetic Acid Water PeroxyChem’s VigorOx® WWT II Wastewater Disinfection Technology contains 15% peracetic acid by weight and 23% hydrogen peroxide as delivered. Although hydrogen peroxide is present in the formulation, peracetic acid is considered to be the active component for disinfection1 in wastewater. There have been several published studies investigating the decomposition kinetics of PAA in different water matrices, including municipal wastewater2-7. Yuan7 states that PAA may be consumed in the following three competitive reactions: 1. Spontaneous decomposition 2 CH3CO3H à 2 CH3CO2H + O2 Eq (1) 2. Hydrolysis CH3CO3H + H2O à CH3CO2H + H2O2 Eq (2) 3. Transition metal catalyzed decomposition + CH3CO3H + M à CH3CO2H + O2 + other products Eq (3) At neutral pH’s, both peracetic acid and hydrogen peroxide can be rapidly consumed by these reactions7 (hydrogen peroxide will decompose to water and oxygen via 2H2O2 à 2H2O + O2). -

Peracetic Acid Processing
Peracetic Acid Processing Identification Chemical Name(s): CAS Number: peroxyacetic acid, ethaneperoxic acid 79-21-0 Other Names: Other Codes: per acid, periacetic acid, PAA NIOSH Registry Number: SD8750000 TRI Chemical ID: 000079210 UN/ID Number: UN3105 Summary Recommendation Synthetic / Allowed or Suggested Non-Synthetic: Prohibited: Annotation: Synthetic Allowed (consensus) Allowed only for direct food contact for use in wash water. Allowed as a (consensus) sanitizer on surfaces in contact with organic food. (consensus) From hydrogen peroxide and fermented acetic acid sources only. (Not discussed by processing reviewers--see discussion of source under Crops PAA TAP review.) Characterization Composition: C2H4O3. Peracetic acid is a mixture of acetic acid (CH3COOH) and hydrogen peroxide (H2O2) in an aqueous solution. Acetic acid is the principle component of vinegar. Hydrogen peroxide has been previously recommended by the NOSB for the National List in processing (synthetic, allowed at Austin, 1995). Properties: It is a very strong oxidizing agent and has stronger oxidation potential than chlorine or chlorine dioxide. Liquid, clear, and colorless with no foaming capability. It has a strong pungent acetic acid odor, and the pH is acid (2.8). Specific gravity is 1.114 and weighs 9.28 pounds per gallon. Stable upon transport. How Made: Peracetic acid (PAA) is produced by reacting acetic acid and hydrogen peroxide. The reaction is allowed to continue for up to ten days in order to achieve high yields of product according to the following equation. O O || || CH3-C-OH + H2O2 CH3C-O-OH + H2O acetic acid hydrogen peroxyacetic peroxide acid Due to reaction limitations, PAA generation can be up to 15% with residual levels of hydrogen peroxide (up to 25%) and acetic acid (up to 35%) with water up to 25%. -

Bio-Solvents: Synthesis, Industrial Production and Applications Novisi K
Chapter Bio-Solvents: Synthesis, Industrial Production and Applications Novisi K. Oklu, Leah C. Matsinha and Banothile C.E. Makhubela Abstract Solvents are at the heart of many research and industrial chemical processes and consumer product formulations, yet an overwhelming number are derived from fossils. This is despite societal and legislative push that more products be produced from carbon-neutral resources, so as to reduce our carbon footprint and environmental impact. Biomass is a promising renewable alternative resource for producing bio-solvents, and this review focuses on their extraction and synthesis on a laboratory and large scale. Starch, lignocellulose, plant oils, animal fats and proteins have been combined with creative synthetic pathways, novel technolo- gies and processes to afford known or new bio-derived solvents including acids, alkanes, aromatics, ionic liquids (ILs), furans, esters, ethers, liquid polymers and deep eutectic solvents (DESs)—all with unique physiochemical properties that warrant their use as solvation agents in manufacturing, pharmaceutical, cosmet- ics, chemicals, energy, food and beverage industries, etc. Selected bio-solvents, conversion technologies and processes operating at commercial and demonstration scale including (1) Solvay’s Augeo™ SL 191 renewable solvent, (2) Circa Group’s Furacell™ technology and process for making levoglucosenone (LGO) to produce dihydrolevoglucosenone (marketed as Cyrene™), (3) Sappi’s Xylex® technology and demonstration scale processes that aim to manufacture precursors for bio- solvents and (4) Anellotech’s Bio-TCat™ technology and process for producing benzene, toluene and xylenes (BTX) are highlighted. Keywords: bio-solvents, renewable resources, green chemistry, biorefinery, biomass 1. Introduction Air quality deterioration, environmental, health and safety issues have raised serious concerns over continued processing of fossil-based feedstocks in producing chemical products such as fuels and solvents. -

Peracetic Acid
Regulation (EU) No 528/2012 concerning the making available on the market and use of biocidal products Evaluation of active substances Assessment Report Peracetic acid Product-types 11 and 12 (Preservatives for liquid cooling and processing systems) (Slimicides) August 2016 Finland Peracetic acid Product-types 11 and 12 August 2016 CONTENTS 1. STATEMENT OF SUBJECT MATTER AND PURPOSE ........................................................ 2 1.1. Procedure followed ............................................................................................. 2 1.2. Purpose of the assessment report ....................................................................... 2 2. OVERALL SUMMARY AND CONCLUSIONS ..................................................................... 4 2.1. Presentation of the Active Substance .................................................................. 4 2.1.1. Identity, Physico-Chemical Properties & Methods of Analysis ................. 4 2.1.2. Intended Uses and Efficacy ..................................................................... 7 2.1.3. Classification and Labelling ..................................................................... 8 2.2. Summary of the Risk Assessment ....................................................................... 9 2.2.1. Human Health Risk Assessment .............................................................. 9 2.2.1.1. Hazard identification ................................................................................... 9 2.2.1.2. Effects assessment ................................................................................... -

Dihydrolevoglucosenone (Cyrene™), a Bio-Based Solvent for Liquid-Liquid Extraction Applications Thomas Brouwer, and Boelo Schuur ACS Sustainable Chem
Subscriber access provided by UNIV TWENTE Article Dihydrolevoglucosenone (Cyrene™), a Bio-based Solvent for Liquid-Liquid Extraction Applications Thomas Brouwer, and Boelo Schuur ACS Sustainable Chem. Eng., Just Accepted Manuscript • DOI: 10.1021/ acssuschemeng.0c04159 • Publication Date (Web): 31 Aug 2020 Downloaded from pubs.acs.org on September 16, 2020 Just Accepted “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts. is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036 Published by American Chemical Society. -
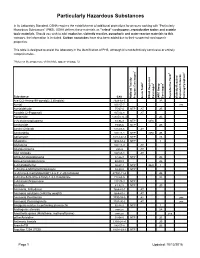
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

Peracetic Acid Bleaching CH CO H
Peracetic Acid Bleaching CH3CO3H Introduction of Bleaching Bleaching is a chemical decoloration and delignification process carried out on various types of pulp. DliDelignifi cati on Removal of the chromophores At in dus tr ia l scal e, bl eachi ng i s perf ormed b y chl ori ne, chl ori ne dioxide, oxygen, hydrogen peroxide, ozone and peracetic acid. Effic iency: ClO2 = Cl2 = O3 >CH3COOOH > H2O2 >O> O2 Often bleaching chemicals are wasted in secondary reactions. D. Lachenal and C. Chirat, Cellulose Chem. Technol., 39, 5-6, 511-156 (2005) 2 Electron Exchange Theory Ring opening of the aromatic units necessitates the exchange of 4 e - # of e- can be exchanged per bleaching molecule - + O3 + 6 e + 6 H → 3 H2O - + - CCOlO2 + 55e e + 4 H → Cl + 2 H2O2 - + O2 + 4 e + 4 H → 2 H2O - + H2O2 + 2 e + 2 H → 2 H2O - + CH3COOOH + 2 e + 2 H → CH3COOH + H2O # of e- exchanged per molecule decreases in the following order: O3 > ClO2 > O2 > H2O2 = CH3COOOH D. Lachenal and C. Chirat, Cellulose Chem. Technol., 39, 5-6, 511-156 (2005) 3 Actual Electron Exchange - ClO2 and O3 100 g un bleac he d SW k raf t pul p, k appa # f rom 30 to ~ 4 -5 - 2.3% ClO2 (0.034 mole) on pulp: 0.034 x 5 = 0.17 e is exchanged - 14%1.4% O3 (0.03 mole)on pulp: 0 .03 x 6 = 018e0.18 e is exchanged Chemicals % consumed1 Kappa number * ClO2 2.3 4.5 O3 1.5 3.5 O3 1.4 5.0 O3 1.3 6.5 1Chemicals consumed are shown as % on pulp. -
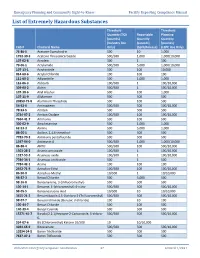
List of Extremely Hazardous Substances
Emergency Planning and Community Right-to-Know Facility Reporting Compliance Manual List of Extremely Hazardous Substances Threshold Threshold Quantity (TQ) Reportable Planning (pounds) Quantity Quantity (Industry Use (pounds) (pounds) CAS # Chemical Name Only) (Spill/Release) (LEPC Use Only) 75-86-5 Acetone Cyanohydrin 500 10 1,000 1752-30-3 Acetone Thiosemicarbazide 500/500 1,000 1,000/10,000 107-02-8 Acrolein 500 1 500 79-06-1 Acrylamide 500/500 5,000 1,000/10,000 107-13-1 Acrylonitrile 500 100 10,000 814-68-6 Acrylyl Chloride 100 100 100 111-69-3 Adiponitrile 500 1,000 1,000 116-06-3 Aldicarb 100/500 1 100/10,000 309-00-2 Aldrin 500/500 1 500/10,000 107-18-6 Allyl Alcohol 500 100 1,000 107-11-9 Allylamine 500 500 500 20859-73-8 Aluminum Phosphide 500 100 500 54-62-6 Aminopterin 500/500 500 500/10,000 78-53-5 Amiton 500 500 500 3734-97-2 Amiton Oxalate 100/500 100 100/10,000 7664-41-7 Ammonia 500 100 500 300-62-9 Amphetamine 500 1,000 1,000 62-53-3 Aniline 500 5,000 1,000 88-05-1 Aniline, 2,4,6-trimethyl- 500 500 500 7783-70-2 Antimony pentafluoride 500 500 500 1397-94-0 Antimycin A 500/500 1,000 1,000/10,000 86-88-4 ANTU 500/500 100 500/10,000 1303-28-2 Arsenic pentoxide 100/500 1 100/10,000 1327-53-3 Arsenous oxide 100/500 1 100/10,000 7784-34-1 Arsenous trichloride 500 1 500 7784-42-1 Arsine 100 100 100 2642-71-9 Azinphos-Ethyl 100/500 100 100/10,000 86-50-0 Azinphos-Methyl 10/500 1 10/10,000 98-87-3 Benzal Chloride 500 5,000 500 98-16-8 Benzenamine, 3-(trifluoromethyl)- 500 500 500 100-14-1 Benzene, 1-(chloromethyl)-4-nitro- 500/500 -

(12) Patent Application Publication (10) Pub. No.: US 2016/0372276 A1 HAN Et Al
US 20160372276A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0372276 A1 HAN et al. (43) Pub. Date: Dec. 22, 2016 (54) A PRECIOUS METAL SWITCH CONTACT C2.5D 5/48 (2006.01) COMPONENT AND ITS PREPARATION C2.5D 3/48 (2006.01) METHOD C2.5D 3/46 (2006.01) C2.5D 5/02 (2006.01) (71) Applicant: NANTONG MEMTECH HIH II/04 (2006.01) TECHNOLOGIES CO., LTD, C23C 8/42 (2006.01) Nantong (CN) (52) U.S. Cl. CPC ................ H01H I/02 (2013.01); H0IH II/04 (72) Inventors: Huisheng HAN, Nantong (CN); (2013.01); C23C 18/1605 (2013.01); C23C HONGMEI ZHANG, NANTONG 18/1689 (2013.01); C23C 18/42 (2013.01); (CN); YANG DING, NANTONG (CN); C25D 3/48 (2013.01); C25D 3/46 (2013.01); ZHIHONG DONG, NANTONG (CN); C25D 5/022 (2013.01); C25D 5/48 (2013.01) CHENG HUANG, NANTONG (CN) (57) ABSTRACT (21) Appl. No.: 14/896,403 This invention discloses a preparation method for precious metal Switching contact components by means of plating (22) PCT Filed: Sep. 15, 2014 masking, plating and etching processes. The plating masking PCT/CN2014/090913 process is performed by using a plating mask ink with or (86). PCT No.: without a photo exposure machine. Plating of precious S 371 (c)(1), metals is performed by electroless plating or electro plating (2) Date: Dec. 7, 2015 methods. Etching is carried out with etching solutions con taining weak organic acids, weak inorganic acids or acidic (30) Foreign Application Priority Data buffering agents. Improvement of the etched Surface gloss and prevention of the side etching are realized with the Sep. -

The Use of Peracetic Acid As a “Pre-Oxidant” for Drinking Water Applications
Disinfection Forum No. 18, September 2017 The Use of Peracetic Acid as a “Pre-Oxidant” for Drinking Water Applications BACKGROUND Chlorination as a technology for drinking water disinfection has significantly reduced the incidence of human disease and is one of the most significant contributions to the improvement of human health over the past century. Chlorine’s ability to provide a stable residual concentration makes it suitable as a drinking water disinfectant at the point of use. However, natural organic matter in the raw water being treated and disinfected at the drinking water treatment plant may interact with the residual chlorine to form compounds classified as disinfection by-products (DBPs). The most commonly employed disinfectants are chlorine, chlorine dioxide and ozone – each of which can generate a variety of DBPs. Many DBPs have been identified as potential cancer-causing agents, and include trihalomethanes (THMs) and haloacetic acids (HAAs). Examples of THMS and HAAs include: chloroform, dibromochloromethane, bromoform, trichloracetic acid, monchloracetic acid and monobromoacetic acid. Peracetic acid (PAA) has gained momentum in the United States as a primary wastewater disinfection technology and alternative to chlorine. PAA has low residual toxicity, does not form harmful DBPs and has minimal impact on the environment. While the lack of a stable residual reduces its environmental impact post wastewater treatment, this characteristic decreases its values as a terminal drinking water disinfection technology – PAA residual at the point of use would be impractical to maintain. However, PAA may potentially be used in a disinfection treatment train approach as an early stage disinfection chemistry coupled with other methods such as membrane filtration, ozone and UV.