2.2.Uncatalysed Formation of Peracetic Acid from Hydrogen Peroxide and Acetic Acid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Decomposition Kinetics of Peracetic Acid and Hydrogen Peroxide in Municipal Wastewaters
Disinfection Forum No 10, October 2015 The Decomposition Kinetics of Peracetic Acid and Hydrogen Peroxide in Municipal Wastewaters INTRODUCTION Efficient control of microbial populations in municipal wastewater using peracetic acid (PAA) requires an understanding of the PAA decomposition kinetics. This knowledge is critical to ensure the proper dosing of PAA needed to achieve an adequate concentration within the contact time of the disinfection chamber. In addition, the impact of PAA on the environment, post-discharge into the receiving water body, also is dependent upon the longevity of the PAA in the environment, before decomposing to acetic acid, oxygen and water. As a result, the decomposition kinetics of PAA may have a significant impact on aquatic and environmental toxicity. PAA is not manufactured as a pure compound. The solution exists as an equilibrium mixture of PAA, hydrogen peroxide, acetic acid, and water: ↔ + + Acetic Acid Hydrogen Peroxide Peracetic Acid Water PeroxyChem’s VigorOx® WWT II Wastewater Disinfection Technology contains 15% peracetic acid by weight and 23% hydrogen peroxide as delivered. Although hydrogen peroxide is present in the formulation, peracetic acid is considered to be the active component for disinfection1 in wastewater. There have been several published studies investigating the decomposition kinetics of PAA in different water matrices, including municipal wastewater2-7. Yuan7 states that PAA may be consumed in the following three competitive reactions: 1. Spontaneous decomposition 2 CH3CO3H à 2 CH3CO2H + O2 Eq (1) 2. Hydrolysis CH3CO3H + H2O à CH3CO2H + H2O2 Eq (2) 3. Transition metal catalyzed decomposition + CH3CO3H + M à CH3CO2H + O2 + other products Eq (3) At neutral pH’s, both peracetic acid and hydrogen peroxide can be rapidly consumed by these reactions7 (hydrogen peroxide will decompose to water and oxygen via 2H2O2 à 2H2O + O2). -

BACKGROUND DOCUMENT for DYES METABOLIZED to BENZIDINE (BENZIDINE DYE CLASS)
NTP REPORT ON CARCINOGENS BACKGROUND DOCUMENT for DYES METABOLIZED TO BENZIDINE (BENZIDINE DYE CLASS) FINAL MARCH1999 Prepared for the October 30-31, 1997, Meeting ofthe Report on Carcinogens Subcommittee ofthe NTP Board ofScientific Counselors Prepared by Integrated Laboratory Systems Post Office Box 13501 Research Triangle Park, North Carolina 27709 NIEHS Contract No. N01-ES-25346 NTP Report on Carcinogens 1997 Background Document for Dyes Metabolized to Benzidine (Benzidine Dye Class) TABLE OF CONTENTS NTP Report on Carcinogens Listing for Dyes Metabolized to Benzidine (Benzidine Dye Class) ...•.......•..•.•.......•...•..•.•.•.••••.•.•.•••.••••••••••••••• 1 Listing Criteria from the Report on Carcinogens, Eighth Edition ••••.••••••.••.•.•.• 3 Supporting Information for Listing .........•...................•.•........•.•.•••.•••.•••.•••.•.••..•... 4 Table 1. Some Regulated Azo Dyes Derived From Benzidine That Have Citations in BIOSIS, CANCERLIT, EMBASE, MEDLINE, RTECS, and/or TOXLINE •.•..•..••..•.••••.•.•.•••.•..•.••••••••••••••••••.•• 5 REFERENCES •.•.•.•.•.••.•.••.•.•.......•..•..•.....•.•..•..•..•.•....•..•..•.•.••••.•.•.••.•••.••••••••••••••.••••• 8 APPENDIX A- Excerpts from IARC (1982a) and IARC Supplements (IARC, 1979; IARC, 1982b; and IARC, 1987) Benzidine .•••...•••.••••••••• A-1 APPENDIX B- Excerpts from IARC (1982a) Direct Black 38, Direct Blue 6, Direct Brown 95 ..•.••.••.••••.••.•••••••.•.••.••••••••.•.•••.•••.•••••••••••••• B-1 APPENDIX C- Excerpts from the NCI Technical Report 13-Week Subchronic Toxicity Studies of Direct -

University of Groningen Discovery of a Eugenol Oxidase From
University of Groningen Discovery of a eugenol oxidase from Rhodococcus sp strain RHA1 Jin, J.F.; Mazon, H.; van den Heuvel, R.H.H.; Janssen, D.B.; Fraaije, M.W. Published in: Febs Journal DOI: 10.1111/j.1742-4658.2007.05767.x IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2007 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Jin, J. F., Mazon, H., van den Heuvel, R. H. H., Janssen, D. B., & Fraaije, M. W. (2007). Discovery of a eugenol oxidase from Rhodococcus sp strain RHA1. Febs Journal, 274(9), 2311 - 2321. https://doi.org/10.1111/j.1742-4658.2007.05767.x Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 23-09-2021 Discovery of a eugenol oxidase from Rhodococcus sp. -

Peracetic Acid Processing
Peracetic Acid Processing Identification Chemical Name(s): CAS Number: peroxyacetic acid, ethaneperoxic acid 79-21-0 Other Names: Other Codes: per acid, periacetic acid, PAA NIOSH Registry Number: SD8750000 TRI Chemical ID: 000079210 UN/ID Number: UN3105 Summary Recommendation Synthetic / Allowed or Suggested Non-Synthetic: Prohibited: Annotation: Synthetic Allowed (consensus) Allowed only for direct food contact for use in wash water. Allowed as a (consensus) sanitizer on surfaces in contact with organic food. (consensus) From hydrogen peroxide and fermented acetic acid sources only. (Not discussed by processing reviewers--see discussion of source under Crops PAA TAP review.) Characterization Composition: C2H4O3. Peracetic acid is a mixture of acetic acid (CH3COOH) and hydrogen peroxide (H2O2) in an aqueous solution. Acetic acid is the principle component of vinegar. Hydrogen peroxide has been previously recommended by the NOSB for the National List in processing (synthetic, allowed at Austin, 1995). Properties: It is a very strong oxidizing agent and has stronger oxidation potential than chlorine or chlorine dioxide. Liquid, clear, and colorless with no foaming capability. It has a strong pungent acetic acid odor, and the pH is acid (2.8). Specific gravity is 1.114 and weighs 9.28 pounds per gallon. Stable upon transport. How Made: Peracetic acid (PAA) is produced by reacting acetic acid and hydrogen peroxide. The reaction is allowed to continue for up to ten days in order to achieve high yields of product according to the following equation. O O || || CH3-C-OH + H2O2 CH3C-O-OH + H2O acetic acid hydrogen peroxyacetic peroxide acid Due to reaction limitations, PAA generation can be up to 15% with residual levels of hydrogen peroxide (up to 25%) and acetic acid (up to 35%) with water up to 25%. -

Chemistry 301-301A - Hour Examination #3, December 11, 2003
Chemistry 301-301A - Hour Examination #3, December 11, 2003 “.....as we know, there are known unknowns; there are things we know we know. We also know there are known unknowns; that is to say we know there are some things we do not know. But there are also unknown unknowns - the ones we don't know we don't know.” Donald Rumsfeld (winner of a British award given to the worst mangler of the English language in 2003) “I know, a proof is a proof. What kind of a proof is a proof? A proof is a proof and when you have a good proof it's because it's proven." Jean Chrétien (hon. mention for the same award) 1[18 points] (a) Acid-catalyzed addition of water to 3-methyl-1-butene (1) results in formation of large amounts of a rearranged alcohol (2), in addition to the expected alcohol (3). Explain, with excellent arrow formalisms. H2O + H O+ 3 OH OH 1 3 2 (b) On the other hand hydroboration of 1, followed by oxidation, does not lead to any rearranged product. Only alcohol 4 is formed. Explain. Detailed mechanisms are not needed here, but a drawing of the transition state for the hydroboration step is. 1. BH3 OH 2. HOOH/HO – 1 4 (c) But there are some strange things that happen in hydroboration. For example when 2-methyl-2-butene (5) is hydroborated at high temperature, then treated with HOOH/HO–, alcohol 4 is still one of the products. Explain mechanistcally. Hint: at high temperature hydroboration is reversible. -

Algorithmic Analysis of Chemical Dynamics of the Autoignition of NH3–H2O2/Air Mixtures
energies Article Algorithmic Analysis of Chemical Dynamics of the Autoignition of NH3–H2O2/Air Mixtures Ahmed T. Khalil 1,2, Dimitris M. Manias 3, Efstathios-Al. Tingas 4, Dimitrios C. Kyritsis 1,2,* and Dimitris A. Goussis 1,2 1 Department of Mechanical Engineering, Khalifa University of Science and Technology, Abu Dhabi 127788, UAE; [email protected] (A.T.K.); [email protected] (D.A.G.) 2 Research and Innovation Center on CO2 and H2 (RICH), Khalifa University of Science and Technology, P.O. Box 127788, Abu Dhabi, UAE 3 Department of Mechanics, School of Applied Mathematics and Physical Sciences, National Technical University of Athens, 157 73 Athens, Greece; [email protected] 4 Perth College, University of the Highlands and Islands, (UHI), Perth PH1 2NX, UK; [email protected] * Correspondence: [email protected] Received: 8 September 2019; Accepted: 7 October 2019; Published: 21 November 2019 Abstract: The dynamics of a homogeneous adiabatic autoignition of an ammonia/air mixture at constant volume was studied, using the algorithmic tools of Computational Singular Perturbation. Since ammonia combustion is characterized by both unrealistically long ignition delays and elevated NOx emissions, the time frame of action of the modes that are responsible for ignition was analyzed by calculating the developing time scales throughout the process and by studying their possible relation to NOx emissions. The reactions that support or oppose the explosive time scale were identified, along with the variables that are related the most to the dynamics that drive the system to an explosion. -

Interaction of Ozone and Hydrogen Peroxide in Water Implications For
UC Irvine Faculty Publications Title Interaction of ozone and hydrogen peroxide in water: Implications for analysis of H 2 O 2 in air Permalink https://escholarship.org/uc/item/7j7454z7 Journal Geophysical Research Letters, 9(3) ISSN 00948276 Authors Zika, R. G Saltzman, E. S Publication Date 1982-03-01 DOI 10.1029/GL009i003p00231 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California GEOPHYSICLARESEARCH LETTERS, VOL. 9, NO. 3, PAGES231-234 , MARCH1982 INTERACTION OF OZONE AND HYDROGEN PEROXIDE IN WATER: IMPLICATIONSFORANALYSIS oFH20 2 IN AIR R.G. Zika and E.S. Saltzman Division of Marine and Atmospheric Chemistry, University of Miami, Miami, Florida 331#9 Abstract. We have attempted to measure gaseous Analytical Methods H202 in air usingan aqueoustrapping method. With continuousbubbling, H 20 2 levels in the traps reacheda a. Hydrogen Peroxide. Hydrogen peroxide in aqueous plateau, indicating that a state of dynamic equilibrium solution was measured using a modified fluorescence involving H202 destrbction was established. We decay technique [Perschke and Broda, 1976; Zika and attribute this behavior to the interaction of ozone and its Zelmer, 1982]. The method involved the addition of a decompositionproducts (OH, O[) withH 20 2 inacld:•ous known amot•at of scopoletin (6-methyl-7-hydroxyl-i,2- solution. This hypothesis was investigated by replacing benzopyrone)to a pH 7.0 phosphatebuffered. sample. the air stream with a mixture of N2, 02 and 0 3. The The sample was prepared bY diluting an aliquot of the results Of this experiment show that H O was both reaction solution to 20 mls with low contaminant producedand destroyedin the traps. -

Peracetic Acid
Regulation (EU) No 528/2012 concerning the making available on the market and use of biocidal products Evaluation of active substances Assessment Report Peracetic acid Product-types 11 and 12 (Preservatives for liquid cooling and processing systems) (Slimicides) August 2016 Finland Peracetic acid Product-types 11 and 12 August 2016 CONTENTS 1. STATEMENT OF SUBJECT MATTER AND PURPOSE ........................................................ 2 1.1. Procedure followed ............................................................................................. 2 1.2. Purpose of the assessment report ....................................................................... 2 2. OVERALL SUMMARY AND CONCLUSIONS ..................................................................... 4 2.1. Presentation of the Active Substance .................................................................. 4 2.1.1. Identity, Physico-Chemical Properties & Methods of Analysis ................. 4 2.1.2. Intended Uses and Efficacy ..................................................................... 7 2.1.3. Classification and Labelling ..................................................................... 8 2.2. Summary of the Risk Assessment ....................................................................... 9 2.2.1. Human Health Risk Assessment .............................................................. 9 2.2.1.1. Hazard identification ................................................................................... 9 2.2.1.2. Effects assessment ................................................................................... -

The Mechanism of the Baeyer–Villiger Rearrangement: Quantum Chemistry and TST Study Supported by Experimental Kinetic Data†
PAPER www.rsc.org/obc | Organic & Biomolecular Chemistry The mechanism of the Baeyer–Villiger rearrangement: quantum chemistry and TST study supported by experimental kinetic data† J. Raul Alvarez-Idaboy,*a Lino Reyesa and Nelaine Mora-Diezb Received 15th August 2007, Accepted 19th September 2007 First published as an Advance Article on the web 2nd October 2007 DOI: 10.1039/b712608e The mechanism of the Baeyer–Villiger rearrangement is modelled for the reaction of propanone with trifluoroperacetic acid, catalyzed by trifluoroacetic acid in dichloromethane, using three DFT methods (B3LYP, BH&HLYP and MPWB1K) and MP2. These results are refined and used to calculate the overall reaction rate coefficient using conventional Transition State Theory. The excellent agreement between the calculated (1.00 × 10−3 L mol−1 s−1) and the experimental (1.8 × 10−3 L mol−1 s−1)rate coefficients at the MPWB1K level strongly supports the mechanism recently proposed by our group. This DFT method is then used to study the mechanism of a larger system: cyclohexanone + trifluoroperacetic acid, for which a very good agreement between the calculated and the experimental rate coefficients is also found (1.37 and 0.32 L mol−1 s−1, respectively). The modelled mechanism is not ionic but neutral, and consists of two concerted steps. The first one is strongly catalyzed while the second one, the migration step, seems not to be catalyzed for the systems under study. The results of this work could be of interest for understanding other reactions in non-polar solvents for which ionic mechanisms have been assumed. -
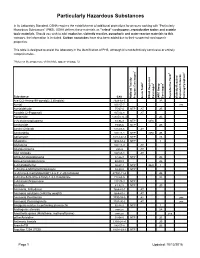
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

Some Aspects the Baeyer-Villicer Reaction
SOME ASPECTS THE BAEYER-VILLICER REACTION - by - JOHN EDVARD BOLLIG-ER. B.So, CSyd.) A. Thesis submitted for the degree of MASTER OF SCIENCE - at the - UNIVERSITY OP NEW SOUTH WALES April, 1963- CONTENTS Page No. Summary . .. 1 The Baeyer-Villiger Reaction .. 2 Discussion: .. .. 26 Baeyer-Villiger Oxidation of 2-Bromocholestan-3-one 32 Baeyer-Villiger Oxidation of 2-Bromofriedelin 50 Baeyer-Villiger Oxidation of 2-Chlorocholestan-3-one 53 Baeyer-Villiger Oxidation of 2-Iodocholestan-3-one 54 Baeyer-Villiger Oxidation of Gerin 56 Experimental .. .. 61 Acknowledgments .. .. 87 Bibliography .. .. 88 1 SUMMARY V/ith a view to synthesising suitable inter mediates for intramolecular Darzen's glycidic ester syntheses, certain steroid and triterpenoid a- substituted ketones have been subjected to Baeyer- Yilliger oxidation. In some cases the oxidation yielded unexpected products and possible mechanisms for their formation are discussed. In other cases the expected products were obtained but readily under went an unusual rearrangement. The structures of the rearranged products have been chemically elucidated and the mechanism of the rearrangement is discussed. This thesis is prefaced by a discussion of the Baeyer-Villiger reaction. THE BAEYER-VTLLIGER REACTION The reaction of ketones with peracids to give esters was first observed by Baeyer and Villiger in 1899 . The scope of this reaction, now known as the Baeyer-Yilliger reaction, has since been widely extended and it has found many useful applications in organic chemistry. Among its representative uses, illustrated by many examples in p the literature , are the formation of esters from simple aliphatic or aromatic ketones, formate esters from aldehydes, anhydrides from o—diketones, lactones from alicyclic ketones and enol lactones from a,0-unsaturated alicyclic ketones. -

Synthesis of an Unsymmetrically Pentafunctionalized Corannulene Derivative (Part I) Synthesis of Platinum and Ethynyl-Platinum Corannulenes (Part II)
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2012 Synthesis of an Unsymmetrically Pentafunctionalized Corannulene Derivative (Part I) Synthesis of Platinum and Ethynyl-Platinum Corannulenes (Part II) Maag, Roman M Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-164179 Dissertation Published Version Originally published at: Maag, Roman M. Synthesis of an Unsymmetrically Pentafunctionalized Corannulene Derivative (Part I) Synthesis of Platinum and Ethynyl-Platinum Corannulenes (Part II). 2012, University of Zurich, Faculty of Science. Part I: Synthesis of an Unsymmetrically Pentafunctionalized Corannulene Derivative and Part II: Synthesis of Platinum and Ethynyl-Platinum Corannulenes Dissertation zur Erlangung der naturwissenschaftlichen Doktorwurde¨ Dr. sc. nat. vorgelegt der Mathematisch-naturwissenschaftlichen Fakult¨at der Universit¨at Zurich¨ von Roman M. Maag von Winkel ZH Promotionskommitee: Prof. Dr. Jay S. Siegel (Vorsitz) Prof. Dr. Kim K. Baldridge Prof. Dr. Cristina Nevado Prof. Dr. Roger Alberto Zurich,¨ 2012 Abstract of the Dissertation Part I: Synthesis of an Unsymmetrically Pentafunctionalized Corannulene Derivative and Part II: Synthesis of Platinum and Ethynyl-Platinum Corannulenes by Roman M. Maag University of Zurich, 2012 Prof. Dr. Jay S. Siegel, Chair Corannulene (C20H10) is a polyaromatic hydrocarbon that can be considered as the smallest fragment of Buckminsterfullerene exhibiting a curved surface. Among the in- teresting properties of corannulene are rapid bowl inversion and esthetically appealing fivefold symmetry (C5v), which is rare in chemistry. Whereas the first synthesis in 1968 only afforded milligram quantities, several improvements in the synthetic strategy finally culminated in the development of an efficient process which today furnishes corannulene in kilogram quantities.