Arabidopsis Thaliana Sang-Hoon Kim1, Claus-Peter Witte2 and Sangkee Rhee 1,3,*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Kinetic Analysis of Pseudouridine Formation in Trna by Archaeal Cbf5 and Pus10
KINETIC ANALYSIS OF PSEUDOURIDINE FORMATION IN TRNA BY ARCHAEAL CBF5 AND PUS10 RAJASHEKHAR KAMALAMPETA M.Sc. University of Skövde, 2008 A Thesis Submitted to the School of Graduate Studies of the University of Lethbridge in Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY IN BIOMOLECULAR SCIENCE Department of Chemistry and Biochemistry University of Lethbridge LETHBRIDGE, ALBERTA, CANADA © Rajashekhar Kamalampeta, 2013 Dedicated to My parents for their unconditional love and support & Those passionate teachers for instilling my interest in science iii Abstract Pseudouridines (), the most common modifications in RNA, are formed by stand-alone synthases in all organisms. In addition, archaea and eukaryotes use H/ACA small ribonucleoproteins for pseudouridylation. Cbf5, the catalytic component of these complexes, can also introduce 55 in archaeal tRNAs in a guide RNA-independent manner. Here, kinetic and thermodynamic analyses revealed that both Pyrococcus furiosus Nop10 and Gar1 proteins enhance the catalytic ability of Cbf5 and increase its affinity for tRNA. Pus10, representing a novel synthase family, is the in vivo archaeal tRNA 55 synthase. Characterization of several Pus10 variants demonstrated the importance of the thumb loop for catalysis, a potential role of the THUMP domain in tRNA binding and a new catalytic arginine which may flip the target uridine into Pus10’s active site. The quantitative characterization of the archaeal pseudouridine synthases Cbf5 and Pus10 reported here sheds light on their cellular roles in RNA modification. iv Acknowledgements First and foremost, I would like to express my sincere gratitude to Dr. Ute Kothe, my supervisor, for her invaluable guidance, mentorship, encouragement, and constructive suggestions throughout my Ph.D. -

Supplemental Table 7. Every Significant Association
Supplemental Table 7. Every significant association between an individual covariate and functional group (assigned to the KO level) as determined by CPGLM regression analysis. Variable Unit RelationshipLabel See also CBCL Aggressive Behavior K05914 + CBCL Emotionally Reactive K05914 + CBCL Externalizing Behavior K05914 + K15665 K15658 CBCL Total K05914 + K15660 K16130 KO: E1.13.12.7; photinus-luciferin 4-monooxygenase (ATP-hydrolysing) [EC:1.13.12.7] :: PFAMS: AMP-binding enzyme; CBQ Inhibitory Control K05914 - K12239 K16120 Condensation domain; Methyltransferase domain; Thioesterase domain; AMP-binding enzyme C-terminal domain LEC Family Separation/Social Services K05914 + K16129 K16416 LEC Poverty Related Events K05914 + K16124 LEC Total K05914 + LEC Turmoil K05914 + CBCL Aggressive Behavior K15665 + CBCL Anxious Depressed K15665 + CBCL Emotionally Reactive K15665 + K05914 K15658 CBCL Externalizing Behavior K15665 + K15660 K16130 KO: K15665, ppsB, fenD; fengycin family lipopeptide synthetase B :: PFAMS: Condensation domain; AMP-binding enzyme; CBCL Total K15665 + K12239 K16120 Phosphopantetheine attachment site; AMP-binding enzyme C-terminal domain; Transferase family CBQ Inhibitory Control K15665 - K16129 K16416 LEC Poverty Related Events K15665 + K16124 LEC Total K15665 + LEC Turmoil K15665 + CBCL Aggressive Behavior K11903 + CBCL Anxiety Problems K11903 + CBCL Anxious Depressed K11903 + CBCL Depressive Problems K11903 + LEC Turmoil K11903 + MODS: Type VI secretion system K01220 K01058 CBCL Anxiety Problems K11906 + CBCL Depressive -

Kinetic Analysis of Pseudouridine Formation in Trna by Archaeal Cbf5 and Pus10
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by OPUS: Open Uleth Scholarship - University of Lethbridge Research Repository University of Lethbridge Research Repository OPUS http://opus.uleth.ca Theses Arts and Science, Faculty of 2013 Kinetic analysis of pseudouridine formation in tRNA by archaeal Cbf5 and Pus10 Kamalampeta, Rajashekhar Lethbridge, Alta. : University of Lethbridge, Dept. of Chemistry and Biochemistry http://hdl.handle.net/10133/3464 Downloaded from University of Lethbridge Research Repository, OPUS KINETIC ANALYSIS OF PSEUDOURIDINE FORMATION IN TRNA BY ARCHAEAL CBF5 AND PUS10 RAJASHEKHAR KAMALAMPETA M.Sc. University of Skövde, 2008 A Thesis Submitted to the School of Graduate Studies of the University of Lethbridge in Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY IN BIOMOLECULAR SCIENCE Department of Chemistry and Biochemistry University of Lethbridge LETHBRIDGE, ALBERTA, CANADA © Rajashekhar Kamalampeta, 2013 Dedicated to My parents for their unconditional love and support & Those passionate teachers for instilling my interest in science iii Abstract Pseudouridines (), the most common modifications in RNA, are formed by stand-alone synthases in all organisms. In addition, archaea and eukaryotes use H/ACA small ribonucleoproteins for pseudouridylation. Cbf5, the catalytic component of these complexes, can also introduce 55 in archaeal tRNAs in a guide RNA-independent manner. Here, kinetic and thermodynamic analyses revealed that both Pyrococcus furiosus Nop10 and Gar1 proteins enhance the catalytic ability of Cbf5 and increase its affinity for tRNA. Pus10, representing a novel synthase family, is the in vivo archaeal tRNA 55 synthase. Characterization of several Pus10 variants demonstrated the importance of the thumb loop for catalysis, a potential role of the THUMP domain in tRNA binding and a new catalytic arginine which may flip the target uridine into Pus10’s active site. -

Within-Host Diversity of Multidrug-Resistant Escherichia Coli in the Gut and Bladder Sofiya G. Shevchenko a Dissertation Submitt
Within-host Diversity of Multidrug-Resistant Escherichia coli in the Gut and Bladder Sofiya G. Shevchenko A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2019 Reading committee: Evgeni Sokurenko, Chair John Mittler Kevin Hybiske Program Authorized to Offer Degree: Microbiology ©Copyright 2019 Sofiya G. Shevchenko University of Washington Abstract Within-host Diversity of Escherichia coli in the Gut and Bladder Sofiya G. Shevchenko Chair of the Supervisory Committee: Evgeni V. Sokurenko Department of Microbiology Uropathogenic E. coli are paradoxically able to both cause disease in the urinary tract, and reside there asymptomatically. The pandemic, multi-drug resistant E. coli subclone ST131-H30 (H30) is of special interest, as it has been found to persist in the gut and bladder of healthy people. In order to understand this persistence, we investigated whether H30 is competitive in these niches and thus able to persist by excluding other E. coli, as well as whether H30 may persist via within-host adaptation. In order to assess the E. coli clonal landscape, we developed a novel method based on deep sequencing of two loci, along with an algorithm for analysis of resulting data. Using this method, we assessed fecal and urinary samples from healthy women carrying H30, and found that even in the absence of antibiotic use, H30 could completely dominate the gut and, especially, urine of healthy carriers. In order to ascertain whether H30 adapts within- host, we employed population-level whole genome sequencing, and determined that H30 undergoes changes in genes affecting respiration in the gut, similar to commensal gut E. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

The HDHD1 Gene, Often Deleted in X-Linked Ichthyosis, Encodes a Pseudouridine-5’-Phosphatase Alice Preumont, Rim Rzem, Didier Vertommen, Emile Van Schaftingen
The HDHD1 gene, often deleted in X-linked ichthyosis, encodes a pseudouridine-5’-phosphatase Alice Preumont, Rim Rzem, Didier Vertommen, Emile van Schaftingen To cite this version: Alice Preumont, Rim Rzem, Didier Vertommen, Emile van Schaftingen. The HDHD1 gene, often deleted in X-linked ichthyosis, encodes a pseudouridine-5’-phosphatase. Biochemical Journal, Portland Press, 2010, 431 (2), pp.237-244. 10.1042/BJ20100174. hal-00521555 HAL Id: hal-00521555 https://hal.archives-ouvertes.fr/hal-00521555 Submitted on 28 Sep 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biochemical Journal Immediate Publication. Published on 20 Aug 2010 as manuscript BJ20100174 The HDHD1 gene, often deleted in X-linked ichthyosis, encodes a pseudouridine-5’- phosphatase Alice Preumont, Rim Rzem, Didier Vertommen, Emile Van Schaftingen From the de Duve Institute, Université Catholique de Louvain, B-1200 Brussels, Belgium Address correspondence to: Emile Van Schaftingen, de Duve Institute, Université catholique de Louvain, Avenue Hippocrate 75, B-1200 Brussels, Belgium. Tel.: 3227647564; Fax: 3227647598; E-mail: [email protected] The abbreviations used are: 5’-PsiMP, pseudouridine 5’-phosphate; 5’-PsiMPase, pseudouridine-5’-phosphatase; DTT, dithiothreitol; FMNPase, FMN phosphatase ; LB, Luria- Bertani. -

(12) Patent Application Publication (10) Pub. No.: US 2012/0266329 A1 Mathur Et Al
US 2012026.6329A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0266329 A1 Mathur et al. (43) Pub. Date: Oct. 18, 2012 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/10 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/24 (2006.01) CI2N 9/02 (2006.01) (75) Inventors: Eric J. Mathur, Carlsbad, CA CI2N 9/06 (2006.01) (US); Cathy Chang, San Marcos, CI2P 2L/02 (2006.01) CA (US) CI2O I/04 (2006.01) CI2N 9/96 (2006.01) (73) Assignee: BP Corporation North America CI2N 5/82 (2006.01) Inc., Houston, TX (US) CI2N 15/53 (2006.01) CI2N IS/54 (2006.01) CI2N 15/57 2006.O1 (22) Filed: Feb. 20, 2012 CI2N IS/60 308: Related U.S. Application Data EN f :08: (62) Division of application No. 1 1/817,403, filed on May AOIH 5/00 (2006.01) 7, 2008, now Pat. No. 8,119,385, filed as application AOIH 5/10 (2006.01) No. PCT/US2006/007642 on Mar. 3, 2006. C07K I4/00 (2006.01) CI2N IS/II (2006.01) (60) Provisional application No. 60/658,984, filed on Mar. AOIH I/06 (2006.01) 4, 2005. CI2N 15/63 (2006.01) Publication Classification (52) U.S. Cl. ................... 800/293; 435/320.1; 435/252.3: 435/325; 435/254.11: 435/254.2:435/348; (51) Int. Cl. 435/419; 435/195; 435/196; 435/198: 435/233; CI2N 15/52 (2006.01) 435/201:435/232; 435/208; 435/227; 435/193; CI2N 15/85 (2006.01) 435/200; 435/189: 435/191: 435/69.1; 435/34; CI2N 5/86 (2006.01) 435/188:536/23.2; 435/468; 800/298; 800/320; CI2N 15/867 (2006.01) 800/317.2: 800/317.4: 800/320.3: 800/306; CI2N 5/864 (2006.01) 800/312 800/320.2: 800/317.3; 800/322; CI2N 5/8 (2006.01) 800/320.1; 530/350, 536/23.1: 800/278; 800/294 CI2N I/2 (2006.01) CI2N 5/10 (2006.01) (57) ABSTRACT CI2N L/15 (2006.01) CI2N I/19 (2006.01) The invention provides polypeptides, including enzymes, CI2N 9/14 (2006.01) structural proteins and binding proteins, polynucleotides CI2N 9/16 (2006.01) encoding these polypeptides, and methods of making and CI2N 9/20 (2006.01) using these polynucleotides and polypeptides. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

(12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur Et Al
US 20150240226A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur et al. (43) Pub. Date: Aug. 27, 2015 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/16 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/02 (2006.01) CI2N 9/78 (2006.01) (71) Applicant: BP Corporation North America Inc., CI2N 9/12 (2006.01) Naperville, IL (US) CI2N 9/24 (2006.01) CI2O 1/02 (2006.01) (72) Inventors: Eric J. Mathur, San Diego, CA (US); CI2N 9/42 (2006.01) Cathy Chang, San Marcos, CA (US) (52) U.S. Cl. CPC. CI2N 9/88 (2013.01); C12O 1/02 (2013.01); (21) Appl. No.: 14/630,006 CI2O I/04 (2013.01): CI2N 9/80 (2013.01); CI2N 9/241.1 (2013.01); C12N 9/0065 (22) Filed: Feb. 24, 2015 (2013.01); C12N 9/2437 (2013.01); C12N 9/14 Related U.S. Application Data (2013.01); C12N 9/16 (2013.01); C12N 9/0061 (2013.01); C12N 9/78 (2013.01); C12N 9/0071 (62) Division of application No. 13/400,365, filed on Feb. (2013.01); C12N 9/1241 (2013.01): CI2N 20, 2012, now Pat. No. 8,962,800, which is a division 9/2482 (2013.01); C07K 2/00 (2013.01); C12Y of application No. 1 1/817,403, filed on May 7, 2008, 305/01004 (2013.01); C12Y 1 1 1/01016 now Pat. No. 8,119,385, filed as application No. PCT/ (2013.01); C12Y302/01004 (2013.01); C12Y US2006/007642 on Mar. 3, 2006. -

Row Labels Gene Name GLU 1 GLU 2 GLU 3 RF 1 RF 2 RF 3
Row Labels Gene Name GLU 1 GLU 2 GLU 3 RF 1 RF 2 RF 3 Ga0073285_102189 S-layer homology domain-containing protein 1549 1805 1683 1214 1230 1364 Ga0073285_10263 pyruvate-ferredoxin/flavodoxin oxidoreductase 901 1003 950 757 754 738 Ga0073285_14010 acetaldehyde dehydrogenase /alcohol 774 880 805 751 771 823 dehydrogenase AdhE Ga0073285_1166 chaperonin GroEL 663 798 645 563 703 716 Ga0073285_11916 enolase 486 378 459 422 454 412 Ga0073285_11684 elongation factor Tu 426 390 479 453 396 361 Ga0073285_11912 glyceraldehyde 3-phosphate dehydrogenase 443 382 352 417 381 367 Ga0073285_101203 ketol-acid reductoisomerase 465 555 441 251 251 280 Ga0073285_10199 4-hydroxy-3-methylbut-2-enyl diphosphate 361 382 343 314 358 372 reductase Ga0073285_102163 ATP synthase F1 subcomplex beta subunit 256 228 255 241 272 295 Ga0073285_1376 DNA-directed RNA polymerase subunit beta' 195 227 186 155 161 171 Ga0073285_12917 D-3-phosphoglycerate dehydrogenase 237 195 192 142 147 138 Ga0073285_10829 pyruvate carboxylase 185 226 195 141 148 151 Ga0073285_1377 DNA-directed RNA polymerase subunit beta 185 212 179 144 145 157 Ga0073285_11620 NAD(P)-dependent iron-only hydrogenase catalytic 98 91 93 233 227 248 subunit Contaminant_TRYP_BOVIN #N/A 157 200 188 130 150 149 Ga0073285_11681 LSU ribosomal protein L4P 198 217 190 108 122 131 Ga0073285_11561 carbamoyl-phosphate synthase large subunit 184 186 149 141 147 142 Ga0073285_101306 polyribonucleotide nucleotidyltransferase 165 164 147 128 146 162 Ga0073285_102105 inosine-5'-monophosphate dehydrogenase 221 178 182 95 116 -
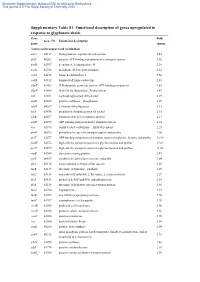
Supplementary Information Table S1. Up-Regulated Known Orfs Induced
Electronic Supplementary Material (ESI) for Molecular BioSystems This journal is © The Royal Society of Chemistry 2012 Supplementary Table S1. Functional description of genes upregulated in response to glyphosate shock Gene Fold Gene ID Functional description name change Amino acid transport and metabolism adiA b4117 biodegradative arginine decarboxylase 2.45 afuC b0262 putative ATP-binding component of a transport system 2.02 ansB b2957 periplasmic L-asparaginase II 2.28 argK b2918 membrane ATPase/protein kinase 2.12 cadA b4131 lysine decarboxylase 1 2.60 cadB b4132 transport of lysine cadaverine 2.83 ddpF b1483 D,D-dipeptide permease system, ATP-binding component 2.83 ddpX b1488 D-ala-D-ala dipeptidase, Zn-dependent 2.49 edd b1851 6-phosphogluconate dehydratase 2.29 elaD b2269 putative sulfatase phosphatase 2.07 idnD b4267 L-idonate dehydrogenase 2.31 nikA b3476 periplasmic binding protein for nickel 2.15 nikB b3477 transport of nickel, membrane protein 2.17 nikD b3479 ATP-binding protein of nickel transport system 2.24 oxc b2373 oxalyl CoA decarboxylase, ThDP-dependent 2.25 pheP b0576 phenylalanine-specific transport system and proline 2.08 proV b2677 ATP-binding component of transport system for glycine, betaine and proline 12.96 proW b2678 high-affinity transport system for glycine betaine and proline 17.69 proX b2679 high-affinity transport system for glycine betaine and proline 11.66 rspB b1580 starvation sensing protein 2.47 speF b0693 ornithine decarboxylase isozyme, inducible 3.04 tdcA b3118 transcriptional activator of tdc operon