Coccidioides Immitis Isolated During Cluster Investigation David M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Coccidioides Immitis
24/08/2017 FUNGAL AGENTS CAUSING INFECTION OF THE LUNG Microbiology Lectures of the Respiratory Diseases Prepared by: Rizalinda Sjahril Microbiology Department Faculty of Medicine Hasanuddin University 2016 OVERVIEW OF CLINICAL MYCOLOGY . Among 150.000 fungi species only 100-150 are human pathogens 25 spp most common pathogens . Majority are saprophyticLiving on dead or decayed organic matter . Transmission Person to person (rare) SPORE INHALATION OR ENTERS THE TISSUE FROM TRAUMA Animal to person (rare) – usually in dermatophytosis 1 24/08/2017 OVERVIEW OF CLINICAL MYCOLOGY . Human is usually resistant to infection, unless: Immunoscompromised (HIV, DM) Serious underlying disease Corticosteroid/antimetabolite treatment . Predisposing factors: Long term intravenous cannulation Complex surgical procedures Prolonged/excessive antibacterial therapy OVERVIEW OF CLINICAL MYCOLOGY . Several fungi can cause a variety of infections: clinical manifestation and severity varies. True pathogens -- have the ability to cause infection in otherwise healthy individuals 2 24/08/2017 Opportunistic/deep mycoses which affect the respiratory system are: Cryptococcosis Aspergillosis Zygomycosis True pathogens are: Blastomycosis Seldom severe Treatment not required unless extensive tissue Coccidioidomycosis destruction compromising respiratory status Histoplasmosis Or extrapulmonary fungal dissemination Paracoccidioidomycosis COMMON PATHOGENS OBTAINED FROM SPECIMENS OF PATIENTS WITH RESPIRATORY DISEASE Fungi Common site of Mode of Infectious Clinical -

Turning on Virulence: Mechanisms That Underpin the Morphologic Transition and Pathogenicity of Blastomyces
Virulence ISSN: 2150-5594 (Print) 2150-5608 (Online) Journal homepage: http://www.tandfonline.com/loi/kvir20 Turning on Virulence: Mechanisms that underpin the Morphologic Transition and Pathogenicity of Blastomyces Joseph A. McBride, Gregory M. Gauthier & Bruce S. Klein To cite this article: Joseph A. McBride, Gregory M. Gauthier & Bruce S. Klein (2018): Turning on Virulence: Mechanisms that underpin the Morphologic Transition and Pathogenicity of Blastomyces, Virulence, DOI: 10.1080/21505594.2018.1449506 To link to this article: https://doi.org/10.1080/21505594.2018.1449506 © 2018 The Author(s). Published by Informa UK Limited, trading as Taylor & Francis Group© Joseph A. McBride, Gregory M. Gauthier and Bruce S. Klein Accepted author version posted online: 13 Mar 2018. Submit your article to this journal Article views: 15 View related articles View Crossmark data Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=kvir20 Publisher: Taylor & Francis Journal: Virulence DOI: https://doi.org/10.1080/21505594.2018.1449506 Turning on Virulence: Mechanisms that underpin the Morphologic Transition and Pathogenicity of Blastomyces Joseph A. McBride, MDa,b,d, Gregory M. Gauthier, MDa,d, and Bruce S. Klein, MDa,b,c a Division of Infectious Disease, Department of Medicine, University of Wisconsin School of Medicine and Public Health, 600 Highland Avenue, Madison, WI 53792, USA; b Division of Infectious Disease, Department of Pediatrics, University of Wisconsin School of Medicine and Public Health, 1675 Highland Avenue, Madison, WI 53792, USA; c Department of Medical Microbiology and Immunology, University of Wisconsin School of Medicine and Public Health, 1550 Linden Drive, Madison, WI 53706, USA. -

Valley Fever a K a Coccidioidomycosis Coccidioidosis Coccidiodal Granuloma San Joaquin Valley Fever Desert Rheumatism Valley Bumps Cocci Cox C
2019 Lung Infection Symposium - Libke 10/26/2019 58 YO ♂ • 1974 PRESENTED WITH HEADACHE – DX = COCCI MENINGITIS WITH HYDROCEPHALUS – Rx = IV AMPHOTERICIN X 6 WKS – VP SHUNT – INTRACISTERNAL AMPHO B X 2.5 YRS (>200 PUNCTURES) • 1978 – 2011 VP SHUNT REVISIONS X 5 • 1974 – 2019 GAINFULLY EMPLOYED, RAISED FAMILY, RETIRED AND CALLS OCCASIONALLY TO SEE HOW I’M DOING. VALLEY FEVER A K A COCCIDIOIDOMYCOSIS COCCIDIOIDOSIS COCCIDIODAL GRANULOMA SAN JOAQUIN VALLEY FEVER DESERT RHEUMATISM VALLEY BUMPS COCCI COX C 1 2019 Lung Infection Symposium - Libke 10/26/2019 COCCIDIOIDOMYCOSIS • DISEASE FIRST DESCRIBED IN 1892 – POSADAS –ARGENTINA – RIXFORD & GILCHRIST - CALIFORNIA – INITIALLY THOUGHT PARASITE – RESEMBLED COCCIDIA “COCCIDIOIDES” – “IMMITIS” = NOT MINOR COCCIDIOIDOMYCOSIS • 1900 ORGANISM IDENTIFIED AS FUNGUS – OPHULS AND MOFFITT – ORGANISM CULTURED FROM TISSUES OF PATIENT – LIFE CYCLE DEFINED – FULFULLED KOCH’S POSTULATES 2 2019 Lung Infection Symposium - Libke 10/26/2019 COCCIDIOIDOMYCOSIS • 1932 ORGANISM IN SOIL SAMPLE FROM DELANO – UNDER BUNKHOUSE OF 4 PATIENTS – DISEASE FATAL • 1937 DICKSON & GIFFORD CONNECTED “VALLEY FEVER” TO C. IMMITIS – USUALLY SELF LIMITED – FREQUENTLY SEEN IN SAN JOAQUIN VALLEY – RESPIRATORY TRACT THE PORTAL OF ENTRY The usual cause for coccidioidomycosis in Arizona is C. immitis A. True B. False 3 2019 Lung Infection Symposium - Libke 10/26/2019 COCCIDIOIDAL SPECIES • COCCIDIOIDES IMMITIS – CALIFORNIA • COCCIDIOIDES POSADASII – NON-CALIFORNIA • ARIZONA, MEXICO • OVERLAP IN SAN DIEGO AREA THE MICROBIAL WORLD • PRIONS -

Valley Fever (Coccidioidomycosis) Tutorial for Primary Care Professionals, Now in Its Second Printing
Valley Fever (Coccidioidomycosis) Tutorial for Primary Care Professionals Prepared by the VALLEY FEVER CENTER FOR EXCELLENCE The University of Arizona 2 TABLE OF CONTENTS Preface ....................................................................................... 4 SECTION 1 ................................................................................. 7 OVERVIEW OF COCCIDIOIDOMYCOSIS History .................................................................................... 8 Mycology ................................................................................ 8 Epidemiology ....................................................................... 10 Spectrum of disease ........................................................... 11 Current therapies ................................................................. 12 SECTION 2 ............................................................................... 13 THE IMPORTANCE OF VALLEY FEVER IN PRIMARY CARE Case reporting ..................................................................... 14 Value of early diagnosis ....................................................... 17 SECTION 3 ............................................................................... 19 PRIMARY CARE MANAGEMENT OF COCCIDIOIDOMYCOSIS C onsider the diagnosis ........................................................ 20 O rder the right tests ............................................................. 23 C heck for risk factors .......................................................... 29 C heck for complications -

Fungal Serology Update – Test Algorithms and Methodologies
LABSTRACT – October 2019 Fungal Serology Update – Test algorithms and methodologies Audience Health care providers and laboratories involved in submission of specimens for fungal serology testing. Overview Beginning October 2019, the fungal serology testing algorithms and methodologies for Aspergillus, Histoplasma capsulatum, Blastomyces dermatitidis and Coccidiodies immitis used at Public Health Ontario have changed. All assays are now Health Canada licensed. Complement fixation (CF) assays have been discontinued. Summary of fungal serology assay methods now in use at PHO Laboratory Serologic Test Method used at PHO Analyte Detected Ordered Aspergillus Enzyme Immunoassay (EIA) IgG antibodies Histoplasma Immunodiffusion M and H band precipitins Blastomyces Immunodiffusion A band precipitins IgM (against TP antigens) and Screen, EIA IgG (against CF antigens) Coccidioides IDTP and IDCF band Confirmation, Immunodiffusion precipitins Page 1 of 4 Fungal Serology Update – Test algorithms and methodologies LAB-SD-134-000 Background information Please note, culture remains the gold standard for the diagnosis of most invasive fungal infections. However, fungal infections can be challenging to diagnose as symptoms are often non-specific and can mimic bacterial, viral, other fungal infections or malignancy. Results of fungal serological tests can be used to aid in the diagnosis and/or management of specific fungal infections and fungal allergies. Aspergillus species are ubiquitous environmental moulds which people inhale each day. While invasive disease is typically restricted to immunocompromised individuals, immunocompetent individuals can have certain allergic manifestations, or develop chronic pulmonary aspergillosis (CPA). Histoplasma capsulatum, Blastomyces dermatitidis and Coccidioides immitis are endemic, dimorphic fungi which can infect otherwise healthy individuals. Most infected individuals are exposed by inhaling fungal spores from the environment and present with initial clinical symptoms of respiratory illness. -
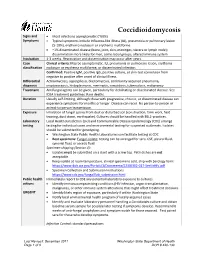
Coccidioidomycosis Reporting and Investigation Guideline
Coccidioidomycosis Signs and • Most infections asymptomatic (~60%) Symptoms • Typical symptoms include influenza-like illness (ILI), pneumonia or pulmonary lesion (5-10%), erythema nodosum or erythema multiforme • ~1% disseminated disease (bone, joint, skin, meninges, viscera or lymph node); dissemination more likely for men, some racial groups, altered immune system Incubation 1-3 weeks. Reactivation and dissemination may occur after years. Case Clinical criteria: May be asymptomatic. ILI, pneumonia or pulmonary lesion, erythema classification nodosum or erythema multiforme, or disseminated infection. Confirmed: Positive IgM, positive IgG, positive culture, or skin-test conversion from negative to positive after onset of clinical illness Differential Actinomycosis, aspergillosis, blastomycosis, community acquired pneumonia, diagnosis cryptococcosis, histoplasmosis, meningitis, sarcoidosis, tuberculosis, malignancy Treatment Antifungal agents can be given, particularly for debilitating or disseminated disease. See IDSA treatment guidelines. Rare deaths. Duration Usually self-limiting, although those with progressive, chronic, or disseminated disease can experience symptoms for months or longer. Disease can recur. No person-to-person or animal-to-person transmission. Exposure Inhalation of fungal spores from dust or disturbed soil (construction, farm work, field training, dust storm, earthquake). Cultures should be handled with BSL2-practices. Laboratory Local Health Jurisdiction (LHJ) and Communicable Disease Epidemiology (CDE) arrange testing testing for individual cases and environmental testing for suspected outbreaks. Isolates should be submitted for genotyping. • Washington State Public Health Laboratories can facilitate testing at CDC • Best specimens: Fungal isolate; testing can be arranged for sera, CSF, pleural fluid, synovial fluid, or ascetic fluid Specimen shipping (Section 4): • Isolates must be submitted on a slant with a screw top. Petri dishes are not acceptable. -

Disseminated Infection Due to Chrysosporium Zonatum in A
JOURNAL OF CLINICAL MICROBIOLOGY, Jan. 1999, p. 18–25 Vol. 37, No. 1 0095-1137/99/$04.0010 Copyright © 1999, American Society for Microbiology. All Rights Reserved. Disseminated Infection Due to Chrysosporium zonatum in a Patient with Chronic Granulomatous Disease and Review of Non-Aspergillus Fungal Infections in Patients with This Disease EMMANUEL ROILIDES,1* LYNNE SIGLER,2 EVANGELIA BIBASHI,3 HELEN KATSIFA,1 4 1 NICOLAS FLARIS, AND CHRISTOS PANTELIADIS Third Department of Pediatrics, Aristotle University of Thessaloniki,1 and Microbiology3 and Pathology Departments,4 Hippokration Hospital, Thessaloniki, Greece, and Microfungus Collection and Herbarium, Devonian Botanic Garden, University of Alberta, Edmonton, Alberta, Canada2 Received 15 June 1998/Returned for modification 1 August 1998/Accepted 6 October 1998 We report the first case of Chrysosporium zonatum infection in a 15-year-old male with chronic granuloma- tous disease who developed a lobar pneumonia and tibia osteomyelitis while on prophylaxis with gamma inter- feron. The fungus was isolated from sputum and affected bone, and hyphae were observed in the bone by his- topathology. Therapy with amphotericin B eradicated the osteomyelitis and pneumonia, but pneumonia recurred in association with pericarditis and pleuritis during therapy with itraconazole. These manifestations subsided, and no recurrences occurred with liposomal amphotericin B therapy. Infections caused by Chrysosporium spe- cies are very rare, and C. zonatum has not previously been reported to cause mycosis in humans. This species, the anamorph of the heterothallic ascomycete Uncinocarpus orissi (family Onygenaceae), is distinguished by its thermotolerance, by colonies which darken from yellowish white to buff, and by club-shaped terminal aleurio- conidia borne at the ends of short, typically curved stalks. -

Coccidioidomycosis in New York State
Synopses Coccidioidomycosis in New York State Vishnu Chaturvedi,* Rama Ramani,* Sally Gromadzki,* Birgit Rodeghier,* Hwa-Gan Chang,† and Dale L. Morse*† New York State Department of Health, Albany, New York, USA; and †School of Public Health, University at Albany, SUNY, Albany, New York, USA Coccidioidomycosis, a systemic fungal disease caused by Coccidioides immitis, is endemic in the southwestern United States and in parts of Mexico and Central and South America. Only sporadic cases have been reported in areas (including New York) where the disease is not endemic. We used hospital discharge records and state mycology laboratory data to investigate the characteristics of C. immitis infections among New York State residents. From 1992 to 1997, 161 persons had hospital discharge diagnoses of coccidioidomycosis (ICD9 Code 114.0 - 114.5, 114.9). From 1989 to 1997, 49 cultures from patients were confirmed as C. immitis; 26 of these patients had traveled to disease-endemic areas. Fourteen of 16 isolates had multilocus genotypes similar to those of Arizona isolates, which corroborates the travel-related acquisition of the disease. Our results indicate that coccidioidomycosis may be more common in New York residents than previously recognized. Increased awareness among health-care providers should improve timely diagnosis of coccidioidomycosis and prevention of associated illnesses and deaths among patients in nondisease- endemic areas. Coccidioidomycosis, a systemic disease which fragment into endospores. When released caused by the dimorphic fungus Coccidioides from the spherule, each endospore can act as a immitis, is endemic in the southwestern United new infectious unit in vivo (1). C. immitis, one of States and parts of Mexico and in Central and the most virulent and infectious fungal South America (1,2). -

Black Fungus: a New Threat Uddin KN
Editorial (BIRDEM Med J 2021; 11(3): 164-165) Black fungus: a new threat Uddin KN Fungal infections, also known as mycoses, are Candida spp. including non-albicans Candida (causing traditionally divided into superficial, subcutaneous and candidiasis), p. Aspergillus spp. (causing aspergillosis), systemic mycoses. Cryptococcus (causing cryptococcosis), Mucormycosis previously called zygomycosis caused by Zygomycetes. What are systemic mycoses? These fungi are found in or on normal skin, decaying Systemic mycoses are fungal infections affecting vegetable matter and bird droppings respectively but internal organs. In the right circumstances, the fungi not exclusively. They are present throughout the world. enter the body via the lungs, through the gut, paranasal sinuses or skin. The fungi can then spread via the Who are at risk of systemic mycoses? bloodstream to multiple organs, often causing multiple Immunocompromised people are at risk of systemic organs to fail and eventually, result in the death of the mycoses. Immunodeficiency can result from: human patient. immunodeficiency virus (HIV) infection, systemic malignancy (cancer), neutropenia, organ transplant What causes systemic mycoses? recipients including haematological stem cell transplant, Patients who are immunocompromised are predisposed after a major surgical operation, poorly controlled to systemic mycoses but systemic mycosis can develop diabetes mellitus, adult-onset immunodeficiency in otherwise healthy patients. Systemic mycoses can syndrome, very old or very young. be split between two main varieties, endemic respiratory infections and opportunistic infections. What are the clinical features of systemic mycoses? The clinical features of a systemic mycosis depend on Endemic respiratory infections the specific infection and which organs have been Fungi that can cause systemic infection in people with affected. -

Neglected Fungal Zoonoses: Hidden Threats to Man and Animals
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REVIEW Neglected fungal zoonoses: hidden threats to man and animals S. Seyedmousavi1,2,3, J. Guillot4, A. Tolooe5, P. E. Verweij2 and G. S. de Hoog6,7,8,9,10 1) Department of Medical Microbiology and Infectious Diseases, Erasmus MC, Rotterdam, 2) Department of Medical Microbiology, Radboud University Medical Centre, Nijmegen, The Netherlands, 3) Invasive Fungi Research Center, Mazandaran University of Medical Sciences, Sari, Iran, 4) Department of Parasitology- Mycology, Dynamyic Research Group, EnvA, UPEC, UPE, École Nationale Vétérinaire d’Alfort, Maisons-Alfort, France, 5) Faculty of Veterinary Medicine, University of Tehran, Tehran, Iran, 6) CBS-KNAW Fungal Biodiversity Centre, Utrecht, 7) Institute for Biodiversity and Ecosystem Dynamics, University of Amsterdam, Amsterdam, The Netherlands, 8) Peking University Health Science Center, Research Center for Medical Mycology, Beijing, 9) Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China and 10) King Abdullaziz University, Jeddah, Saudi Arabia Abstract Zoonotic fungi can be naturally transmitted between animals and humans, and in some cases cause significant public health problems. A number of mycoses associated with zoonotic transmission are among the group of the most common fungal diseases, worldwide. It is, however, notable that some fungal diseases with zoonotic potential have lacked adequate attention in international public health efforts, leading to insufficient attention on their preventive strategies. This review aims to highlight some mycoses whose zoonotic potential received less attention, including infections caused by Talaromyces (Penicillium) marneffei, Lacazia loboi, Emmonsia spp., Basidiobolus ranarum, Conidiobolus spp. -

<I>Chrysosporium</I> Species with Keratinolytic Activity
MYCOTAXON Volume 110, pp. 65–71 October–December 2009 Chrysosporium linfenense: a new Chrysosporium species with keratinolytic activity Jiandong Liang 1,2, Yanfeng Han 2, Wen Du 2, Zongqi Liang2* & Zizhong Li1 [email protected],2 [email protected] [email protected] [email protected] [email protected] 1The Provincial Key Laboratory for Agricultural Pest Management of Mountainous Region, Institute of Entomology , Guizhou University Guiyang, China 550025 2Institute of Fungus Resources, College of Life Sciences, Guizhou University Guiyang, China 550025 Abstract — Chrysosporium linfenense, a new Chrysosporium species, was collected from Shanxi, China, described, and illustrated. Differences between C. linfenense and related species were analyzed based on the morphological and DNA sequence characters. Diagnostic characters of C. linfenense are conidia that are solitary or often in chains of 2–3, mostly ellipsoidal or fusiform, few clavate, and smooth-walled; intercalary conidia are absent. The presence of keratinase also suggests that C. linfenense possesses a keratinolytic activity. Keywords — mitosporic fungi, morphology, molecular analysis, classification Introduction Chrysosporium species distributed around the world can produce many useful metabolites, especially keratinase, which can be used widely in the chemical industry and environmental protection, medical, and agricultural fields (Kushwaha 2000, Liang et al. 2007). The strain studied (GZUIFR-H31) was isolated from the rhizosphere soil of Cedrus deodara. Based on morphological and ITS1-5.8S-ITS2 rDNA sequence characters, this fungus was identified as a new species of Chrysosporium, C. linfenense. Materials and methods Sample collection and strain isolation Strain GZUIFR-H31 was collected and isolated from the rhizosphere soil of Cedrus deodara in Linfen city, Shanxi Province, China. -

Treatment of Fungal Infections in Adult Pulmonary and Critical Care Patients
American Thoracic Society Documents An Official American Thoracic Society Statement: Treatment of Fungal Infections in Adult Pulmonary and Critical Care Patients Andrew H. Limper, Kenneth S. Knox, George A. Sarosi, Neil M. Ampel, John E. Bennett, Antonino Catanzaro, Scott F. Davies, William E. Dismukes, Chadi A. Hage, Kieren A. Marr, Christopher H. Mody, John R. Perfect, and David A. Stevens, on behalf of the American Thoracic Society Fungal Working Group THIS OFFICIAL STATEMENT OF THE AMERICAN THORACIC SOCIETY (ATS) WAS APPROVED BY THE ATS BOARD OF DIRECTORS, MAY 2010 CONTENTS immune-compromised and critically ill patients, including crypto- coccosis, aspergillosis, candidiasis, and Pneumocystis pneumonia; Introduction and rare and emerging fungal infections. Methods Antifungal Agents: General Considerations Keywords: fungal pneumonia; amphotericin; triazole antifungal; Polyenes echinocandin Triazoles Echinocandins The incidence, diagnosis, and clinical severity of pulmonary Treatment of Fungal Infections fungal infections have dramatically increased in recent years in Histoplasmosis response to a number of factors. Growing numbers of immune- Sporotrichosis compromised patients with malignancy, hematologic disease, Blastomycosis and HIV, as well as those receiving immunosupressive drug Coccidioidomycosis regimens for the management of organ transplantation or Paracoccidioidomycosis autoimmune inflammatory conditions, have significantly con- Cryptococcosis tributed to an increase in the incidence of these infections. Aspergillosis Definitive