Gene Expression Profiles Discriminate Between Pathological Complete Response and Resistance to Neoadjuvant FEC100 in Breast Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Original Article Bioinformatic Analysis of Gene Expression Profile in Prostate Epithelial Cells Exposed to Low-Dose Cadmium
Int J Clin Exp Med 2018;11(3):1669-1678 www.ijcem.com /ISSN:1940-5901/IJCEM0062792 Original Article Bioinformatic analysis of gene expression profile in prostate epithelial cells exposed to low-dose cadmium Qiling Liu1,2*, Rongqiang Zhang2*, Xiang Wang1, Peili Wang2, Xiaomei Ren2, Na Sun2, Xiangwen Li2, Xinhui Li2, Chunxu Hai1 1Department of Toxicology, The Ministry of Education Key Lab of Hazard Assessment and Control in Special Op- erational Environment, Shaanxi Provincial Key Lab of Free Radical Biology and Medicine, School of Public Health, Medical University of The Air Force, Xi’an, Shaanxi 710032, China; 2Department of Epidemic and Health Statis- tics, The College of Public Health for The Shaanxi University of Chinese Medicine, Shaanxi 712046, China. *Equal contributors. Received July 25, 2017; Accepted February 5, 2018; Epub March 15, 2018; Published March 30, 2018 Abstract: Objective: This study was to identify key genes and biological pathways involved in responses of prostate epithelial cells after low-dose Cd exposure by using bioinformatic analysis. Methods: The gene chip data of prostate epithelial cells after low-dose Cd exposure were collected from public databases Gene Expression Omnibus. After identification of differentially expressed genes (DEGs), data were input into Qlucore Omics Explorer, Network Analyst, String, and Genclip for further analysis of gene expression profiles, protein-protein interactions (PPI) and protein- chemicals interactions, and critical molecular pathways. Results: A total of 384 DEGs were identified in Cd treated group compared with control group. The number of DEGs gradually decreased over time, with the largest number at 0 h. Furthermore, NDUFB5 (A, S), CYC1, UQCRB, ETFA (B), SNRPD2, and LSM3 (5, 6) were the hub proteins in the PPI network. -

Low Abundance of the Matrix Arm of Complex I in Mitochondria Predicts Longevity in Mice
ARTICLE Received 24 Jan 2014 | Accepted 9 Apr 2014 | Published 12 May 2014 DOI: 10.1038/ncomms4837 OPEN Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice Satomi Miwa1, Howsun Jow2, Karen Baty3, Amy Johnson1, Rafal Czapiewski1, Gabriele Saretzki1, Achim Treumann3 & Thomas von Zglinicki1 Mitochondrial function is an important determinant of the ageing process; however, the mitochondrial properties that enable longevity are not well understood. Here we show that optimal assembly of mitochondrial complex I predicts longevity in mice. Using an unbiased high-coverage high-confidence approach, we demonstrate that electron transport chain proteins, especially the matrix arm subunits of complex I, are decreased in young long-living mice, which is associated with improved complex I assembly, higher complex I-linked state 3 oxygen consumption rates and decreased superoxide production, whereas the opposite is seen in old mice. Disruption of complex I assembly reduces oxidative metabolism with concomitant increase in mitochondrial superoxide production. This is rescued by knockdown of the mitochondrial chaperone, prohibitin. Disrupted complex I assembly causes premature senescence in primary cells. We propose that lower abundance of free catalytic complex I components supports complex I assembly, efficacy of substrate utilization and minimal ROS production, enabling enhanced longevity. 1 Institute for Ageing and Health, Newcastle University, Newcastle upon Tyne NE4 5PL, UK. 2 Centre for Integrated Systems Biology of Ageing and Nutrition, Newcastle University, Newcastle upon Tyne NE4 5PL, UK. 3 Newcastle University Protein and Proteome Analysis, Devonshire Building, Devonshire Terrace, Newcastle upon Tyne NE1 7RU, UK. Correspondence and requests for materials should be addressed to T.v.Z. -

(BPA) Exposure Biomarkers in Ovarian Cancer
Journal of Clinical Medicine Article Identification of Potential Bisphenol A (BPA) Exposure Biomarkers in Ovarian Cancer Aeman Zahra 1, Qiduo Dong 1, Marcia Hall 1,2 , Jeyarooban Jeyaneethi 1, Elisabete Silva 1, Emmanouil Karteris 1,* and Cristina Sisu 1,* 1 Biosciences, College of Health, Medicine and Life Sciences, Brunel University London, Uxbridge UB8 3PH, UK; [email protected] (A.Z.); [email protected] (Q.D.); [email protected] (M.H.); [email protected] (J.J.); [email protected] (E.S.) 2 Mount Vernon Cancer Centre, Northwood HA6 2RN, UK * Correspondence: [email protected] (E.K.); [email protected] (C.S.) Abstract: Endocrine-disrupting chemicals (EDCs) can exert multiple deleterious effects and have been implicated in carcinogenesis. The xenoestrogen Bisphenol A (BPA) that is found in various consumer products has been involved in the dysregulation of numerous signalling pathways. In this paper, we present the analysis of a set of 94 genes that have been shown to be dysregulated in presence of BPA in ovarian cancer cell lines since we hypothesised that these genes might be of biomarker potential. This study sought to identify biomarkers of disease and biomarkers of disease- associated exposure. In silico analyses took place using gene expression data extracted from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx) databases. Differential expression was further validated at protein level using immunohistochemistry on an ovarian cancer tissue microarray. We found that 14 out of 94 genes are solely dysregulated in the presence of BPA, while the remaining 80 genes are already dysregulated (p-value < 0.05) in their expression pattern Citation: Zahra, A.; Dong, Q.; Hall, as a consequence of the disease. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

THE FUNCTIONAL SIGNIFICANCE of MITOCHONDRIAL SUPERCOMPLEXES in C. ELEGANS by WICHIT SUTHAMMARAK Submitted in Partial Fulfillment
THE FUNCTIONAL SIGNIFICANCE OF MITOCHONDRIAL SUPERCOMPLEXES in C. ELEGANS by WICHIT SUTHAMMARAK Submitted in partial fulfillment of the requirements For the degree of Doctor of Philosophy Dissertation Advisor: Drs. Margaret M. Sedensky & Philip G. Morgan Department of Genetics CASE WESTERN RESERVE UNIVERSITY January, 2011 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of _____________________________________________________ candidate for the ______________________degree *. (signed)_______________________________________________ (chair of the committee) ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. Dedicated to my family, my teachers and all of my beloved ones for their love and support ii ACKNOWLEDGEMENTS My advanced academic journey began 5 years ago on the opposite side of the world. I traveled to the United States from Thailand in search of a better understanding of science so that one day I can return to my homeland and apply the knowledge and experience I have gained to improve the lives of those affected by sickness and disease yet unanswered by science. Ultimately, I hoped to make the academic transition into the scholarly community by proving myself through scientific research and understanding so that I can make a meaningful contribution to both the scientific and medical communities. The following dissertation would not have been possible without the help, support, and guidance of a lot of people both near and far. I wish to thank all who have aided me in one way or another on this long yet rewarding journey. My sincerest thanks and appreciation goes to my advisors Philip Morgan and Margaret Sedensky. -

Snps) in Genes of the Alcohol Dehydrogenase, Glutathione S-Transferase, and Nicotinamide Adenine Dinucleotide, Reduced (NADH) Ubiquinone Oxidoreductase Families
J Hum Genet (2001) 46:385–407 © Jpn Soc Hum Genet and Springer-Verlag 2001 ORIGINAL ARTICLE Aritoshi Iida · Susumu Saito · Akihiro Sekine Takuya Kitamoto · Yuri Kitamura · Chihiro Mishima Saori Osawa · Kimie Kondo · Satoko Harigae Yusuke Nakamura Catalog of 434 single-nucleotide polymorphisms (SNPs) in genes of the alcohol dehydrogenase, glutathione S-transferase, and nicotinamide adenine dinucleotide, reduced (NADH) ubiquinone oxidoreductase families Received: March 15, 2001 / Accepted: April 6, 2001 Abstract An approach based on development of a large Introduction archive of single-nucleotide polymorphisms (SNPs) throughout the human genome is expected to facilitate large-scale studies to identify genes associated with drug Human genetic variations result from a dynamic process efficacy and side effects, or susceptibility to common over time that includes sudden mutations, random genetic diseases. We have already described collections of SNPs drift, and, in some cases, founder effects. Variations at a present among various genes encoding drug-metabolizing single gene locus are useful as markers of individual risk for enzymes. Here we report SNPs for such enzymes at adverse drug reactions or susceptibility to complex diseases additional loci, including 8 alcohol dehydrogenases, 12 (for reviews, see Risch and Merikangas 1996; Kruglyak glutathione S-transferases, and 18 belonging to the NADH- 1997; McCarthy and Hilfiker 2000). Common types of ubiquinone oxidoreductase family. Among DNA samples sequence variation in the human genome include single- from 48 Japanese volunteers, we identified a total of 434 nucleotide polymorphisms (SNPs), insertion/deletion SNPs at these 38 loci: 27 within coding elements, 52 in 59 polymorphisms, and variations in the number of repeats of flanking regions, five in 59 untranslated regions, 293 in certain motifs (e.g., microsatellites and variable number of introns, 20 in 39 untranslated regions, and 37 in 39 flanking tandem repeat loci). -
Download Validation Data
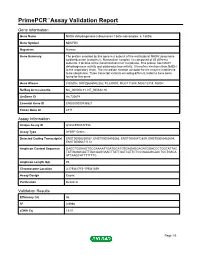
PrimePCR™Assay Validation Report Gene Information Gene Name NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 5, 16kDa Gene Symbol NDUFB5 Organism Human Gene Summary The protein encoded by this gene is a subunit of the multisubunit NADH:ubiquinone oxidoreductase (complex I). Mammalian complex I is composed of 45 different subunits. It locates at the mitochondrial inner membrane. This protein has NADH dehydrogenase activity and oxidoreductase activity. It transfers electrons from NADH to the respiratory chain. The immediate electron acceptor for the enzyme is believed to be ubiquinone. Three transcript variants encoding different isoforms have been found for this gene. Gene Aliases CISGDH, DKFZp686N02262, FLJ30597, MGC111204, MGC12314, SGDH RefSeq Accession No. NC_000003.11, NT_005612.16 UniGene ID Hs.730674 Ensembl Gene ID ENSG00000136521 Entrez Gene ID 4711 Assay Information Unique Assay ID qHsaCED0037586 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000259037, ENST00000493866, ENST00000472629, ENST00000482604, ENST00000471112 Amplicon Context Sequence GAGCTGGAAGTGCGAAAATTGATGCATGTGAGAGGAGATGGACCCTGGTATTAC TATGAGACAATTGACAAGGAACTTATTGATCATTCTCCGAAAGCAACTCCTGACA ATTAAGCATTTTTTTC Amplicon Length (bp) 95 Chromosome Location 3:179341715-179341839 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 96 R2 0.9998 cDNA Cq 19.01 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79 gDNA Cq 24.09 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Electron Transport Chain Activity Is a Predictor and Target for Venetoclax Sensitivity in Multiple Myeloma
ARTICLE https://doi.org/10.1038/s41467-020-15051-z OPEN Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma Richa Bajpai1,7, Aditi Sharma 1,7, Abhinav Achreja2,3, Claudia L. Edgar1, Changyong Wei1, Arusha A. Siddiqa1, Vikas A. Gupta1, Shannon M. Matulis1, Samuel K. McBrayer 4, Anjali Mittal3,5, Manali Rupji 6, Benjamin G. Barwick 1, Sagar Lonial1, Ajay K. Nooka 1, Lawrence H. Boise 1, Deepak Nagrath2,3,5 & ✉ Mala Shanmugam 1 1234567890():,; The BCL-2 antagonist venetoclax is highly effective in multiple myeloma (MM) patients exhibiting the 11;14 translocation, the mechanistic basis of which is unknown. In evaluating cellular energetics and metabolism of t(11;14) and non-t(11;14) MM, we determine that venetoclax-sensitive myeloma has reduced mitochondrial respiration. Consistent with this, low electron transport chain (ETC) Complex I and Complex II activities correlate with venetoclax sensitivity. Inhibition of Complex I, using IACS-010759, an orally bioavailable Complex I inhibitor in clinical trials, as well as succinate ubiquinone reductase (SQR) activity of Complex II, using thenoyltrifluoroacetone (TTFA) or introduction of SDHC R72C mutant, independently sensitize resistant MM to venetoclax. We demonstrate that ETC inhibition increases BCL-2 dependence and the ‘primed’ state via the ATF4-BIM/NOXA axis. Further, SQR activity correlates with venetoclax sensitivity in patient samples irrespective of t(11;14) status. Use of SQR activity in a functional-biomarker informed manner may better select for MM patients responsive to venetoclax therapy. 1 Department of Hematology and Medical Oncology, Winship Cancer Institute, School of Medicine, Emory University, Atlanta, GA, USA. -

A High-Fat Diet Coordinately Downregulates Genes Required for Mitochondrial Oxidative Phosphorylation in Skeletal Muscle Lauren M
A High-Fat Diet Coordinately Downregulates Genes Required for Mitochondrial Oxidative Phosphorylation in Skeletal Muscle Lauren M. Sparks, Hui Xie, Robert A. Koza, Randall Mynatt, Matthew W. Hulver, George A. Bray, and Steven R. Smith Obesity and type 2 diabetes have been associated with a ray studies have shown that genes involved in oxidative high-fat diet (HFD) and reduced mitochondrial mass phosphorylation (OXPHOS) exhibit reduced expression and function. We hypothesized a HFD may affect expres- levels in the skeletal muscle of type 2 diabetic subjects and sion of genes involved in mitochondrial function and prediabetic subjects. These changes may be mediated by biogenesis. To test this hypothesis, we fed 10 insulin- the peroxisome proliferator–activated receptor ␥ coacti- sensitive males an isoenergetic HFD for 3 days with vator-1 (PGC1) pathway. PGC1␣- and PGC1-responsive muscle biopsies before and after intervention. Oligonu- cleotide microarray analysis revealed 297 genes were OXPHOS genes show reduced expression in the muscle of differentially regulated by the HFD (Bonferonni ad- patients with type 2 diabetes (3,4). In addition to the justed P < 0.001). Six genes involved in oxidative cellular energy sensor AMP kinase, the peroxisome prolif- phosphorylation (OXPHOS) decreased. Four were mem- erator–activated receptor cofactors PGC1␣ (5–7) and pos- bers of mitochondrial complex I: NDUFB3, NDUFB5, sibly PGC1 (8) activate mitochondrial biogenesis and NDUFS1, and NDUFV1; one was SDHB in complex II and increase OXPHOS gene expression by increasing the tran- a mitochondrial carrier protein SLC25A12. Peroxisome scription, translation, and activation of the transcription proliferator–activated receptor ␥ coactivator-1 (PGC1) -and PGC1 mRNA were decreased by ؊20%, P < 0.01, factors necessary for mitochondrial DNA (mtDNA) repli ␣ and ؊25%, P < 0.01, respectively. -

Mouse Ndufb5 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Ndufb5 Knockout Project (CRISPR/Cas9) Objective: To create a Ndufb5 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Ndufb5 gene (NCBI Reference Sequence: NM_025316 ; Ensembl: ENSMUSG00000027673 ) is located on Mouse chromosome 3. 6 exons are identified, with the ATG start codon in exon 1 and the TGA stop codon in exon 6 (Transcript: ENSMUST00000127477). Exon 2~5 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 2 starts from about 22.05% of the coding region. Exon 2~5 covers 57.32% of the coding region. The size of effective KO region: ~3714 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 5 6 Legends Exon of mouse Ndufb5 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. So this region is suitable for PCR screening or sequencing analysis. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of Exon 5 is aligned with itself to determine if there are tandem repeats. -

Gene Expression Differences Associated with Human Papillomavirus Status in Head and Neck Squamous Cell Carcinoma Robbertj.C
Human Cancer Biology Gene Expression Differences Associated with Human Papillomavirus Status in Head and Neck Squamous Cell Carcinoma RobbertJ.C. Slebos,1, 2 Yajun Yi, 7 Kim Ely,3 Jesse Carter,8 Amy Evjen,1Xueqiong Zhang,4 Yu Shyr,4 Barbara M. Murphy,8 Anthony J. Cmelak,5 Brian B. Burkey,2 James L. Netterville,2 Shawn Levy,6 Wendell G. Yarbrough,1, 2 and Christine H. Chung8 Abstract Human papillomavirus (HPV) is associated with a subset of head and neck squamous cell carcinoma (HNSCC). Between 15% and 35% of HNSCCs harbor HPV DNA. Demographic and exposure differences between HPV-positive (HPV+) and negative (HPVÀ) HNSCCs suggest that HPV + tumors may constitute a subclass with different biology, whereas clinical differences have also been observed. Gene expression profiles of HPV+ and HPVÀ tumors were compared with further exploration of the biological effect of HPV in HNSCC. Thirty-six HNSCC tumors were analyzed using Affymetrix Human 133U Plus 2.0 GeneChip and for HPV by PCR and real-time PCR. Eight of 36 (22%) tumors were positive for HPV subtype 16. Statistical analysis using Significance Analysis of Microarrays based on HPV status as a supervising variable resulted in a list of 91genes that were differentially expressed with statistical significance. Results for a subset of these genes were verified by real-time PCR. Genes highly expressed in HPV+ samples included cell cycle regulators (p16INK4A, p18, and CDC7) and transcription factors (TAF7L, RFC4, RPA2, andTFDP2).The microarray data were also investigated by mapping genes by chromosomal loca- tion (DIGMAP).A large number of genes on chromosome 3q24-qter had high levels of expression in HPV+ tumors. -

Bayesian Hidden Markov Tree Models for Clustering Genes with Shared Evolutionary History
Submitted to the Annals of Applied Statistics arXiv: arXiv:0000.0000 BAYESIAN HIDDEN MARKOV TREE MODELS FOR CLUSTERING GENES WITH SHARED EVOLUTIONARY HISTORY By Yang Liy,{,∗, Shaoyang Ningy,∗, Sarah E. Calvoz,x,{, Vamsi K. Moothak,z,x,{ and Jun S. Liuy Harvard Universityy, Broad Institutez, Harvard Medical Schoolx, Massachusetts General Hospital{, and Howard Hughes Medical Institutek Determination of functions for poorly characterized genes is cru- cial for understanding biological processes and studying human dis- eases. Functionally associated genes are often gained and lost together through evolution. Therefore identifying co-evolution of genes can predict functional gene-gene associations. We describe here the full statistical model and computational strategies underlying the orig- inal algorithm CLustering by Inferred Models of Evolution (CLIME 1.0) recently reported by us [Li et al., 2014]. CLIME 1.0 employs a mixture of tree-structured hidden Markov models for gene evolution process, and a Bayesian model-based clustering algorithm to detect gene modules with shared evolutionary histories (termed evolutionary conserved modules, or ECMs). A Dirichlet process prior was adopted for estimating the number of gene clusters and a Gibbs sampler was developed for posterior sampling. We further developed an extended version, CLIME 1.1, to incorporate the uncertainty on the evolution- ary tree structure. By simulation studies and benchmarks on real data sets, we show that CLIME 1.0 and CLIME 1.1 outperform traditional methods that use simple metrics (e.g., the Hamming distance or Pear- son correlation) to measure co-evolution between pairs of genes. 1. Introduction. The human genome encodes more than 20,000 protein- coding genes, of which a large fraction do not have annotated function to date [Galperin and Koonin, 2010].