High-Value Oxy-Pharmaceuticals from P450 BM3 ‘Gatekeeper’ Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Title Total Biosynthesis of Opiates by Stepwise Fermentation Using
Total biosynthesis of opiates by stepwise fermentation using Title engineered Escherichia coli. Nakagawa, Akira; Matsumura, Eitaro; Koyanagi, Takashi; Katayama, Takane; Kawano, Noriaki; Yoshimatsu, Kayo; Author(s) Yamamoto, Kenji; Kumagai, Hidehiko; Sato, Fumihiko; Minami, Hiromichi Citation Nature communications (2016), 7 Issue Date 2016-02-05 URL http://hdl.handle.net/2433/204378 This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; Right if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ Type Journal Article Textversion publisher Kyoto University ARTICLE Received 30 Oct 2015 | Accepted 7 Dec 2015 | Published 5 Feb 2016 DOI: 10.1038/ncomms10390 OPEN Total biosynthesis of opiates by stepwise fermentation using engineered Escherichia coli Akira Nakagawa1, Eitaro Matsumura1, Takashi Koyanagi1, Takane Katayama2, Noriaki Kawano3, Kayo Yoshimatsu3, Kenji Yamamoto1, Hidehiko Kumagai1, Fumihiko Sato4 & Hiromichi Minami1 Opiates such as morphine and codeine are mainly obtained by extraction from opium poppies. Fermentative opiate production in microbes has also been investigated, and complete biosynthesis of opiates from a simple carbon source has recently been accom- plished in yeast. Here we demonstrate that Escherichia coli serves as an efficient, robust and flexible platform for total opiate synthesis. Thebaine, the most important raw material in opioid preparations, is produced by stepwise culture of four engineered strains at yields of 2.1 mg l À 1 from glycerol, corresponding to a 300-fold increase from recently developed yeast systems. -

“Biosynthesis of Morphine in Mammals”
“Biosynthesis of Morphine in Mammals” D i s s e r t a t i o n zur Erlangung des akademischen Grades Doctor rerum naturalium (Dr. rer. nat.) vorgelegt der Naturwissenschaftlichen Fakultät I Biowissenschaften der Martin-Luther-Universität Halle-Wittenberg von Frau Nadja Grobe geb. am 21.08.1981 in Querfurt Gutachter /in 1. 2. 3. Halle (Saale), Table of Contents I INTRODUCTION ........................................................................................................1 II MATERIAL & METHODS ........................................................................................ 10 1 Animal Tissue ....................................................................................................... 10 2 Chemicals and Enzymes ....................................................................................... 10 3 Bacteria and Vectors ............................................................................................ 10 4 Instruments ........................................................................................................... 11 5 Synthesis ................................................................................................................ 12 5.1 Preparation of DOPAL from Epinephrine (according to DUNCAN 1975) ................. 12 5.2 Synthesis of (R)-Norlaudanosoline*HBr ................................................................. 12 5.3 Synthesis of [7D]-Salutaridinol and [7D]-epi-Salutaridinol ..................................... 13 6 Application Experiments ..................................................................................... -

Dorset Medicines Advisory Group
DORSET CARDIOLOGY WORKING GROUP GUIDELINE FOR CALCIUM CHANNEL BLOCKERS IN HYPERTENSION SUMMARY The pan-Dorset cardiology working group continues to recommend the use of amlodipine (a third generation dihydropyridine calcium-channel blocker) as first choice calcium channel blocker on the pan-Dorset formulary for hypertension. Lercanidipine is second choice, lacidipine third choice and felodipine is fourth choice. This is due to preferable side effect profiles in terms of ankle oedema and relative costs of the preparations. Note: where angina is the primary indication or is a co-morbidity prescribers must check against the specific product characteristics (SPC) for an individual drug to confirm this is a licensed indication. N.B. Lacidipine and lercandipine are only licensed for use in hypertension. Chapter 02.06.02 CCBs section of the Formulary has undergone an evidence-based review. A comprehensive literature search was carried out on NHS Evidence, Medline, EMBASE, Cochrane Database, and UK Duets. This was for recent reviews or meta-analyses on calcium channel blockers from 2009 onwards (comparative efficacy and side effects) and randomised controlled trials (RCTs). REVIEW BACKGROUND Very little good quality evidence exists. No reviews, meta-analyses or RCTs were found covering all calcium channel blockers currently on the formulary. Another limitation was difficulty obtaining full text original papers for some of the references therefore having to use those from more obscure journals instead. Some discrepancies exist between classification of generations of dihydropyridine CCBs, depending upon the year of publication of the reference/authors’ interpretation. Dihydropyridine (DHP) CCBs tend to be more potent vasodilators than non-dihydropyridine (non-DHP) CCBs (diltiazem, verapamil), but the latter have greater inotropic effects. -
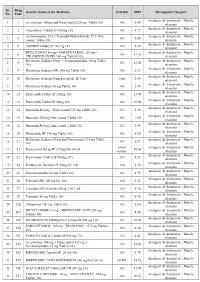
PMBJP Product.Pdf
Sr. Drug Generic Name of the Medicine Unit Size MRP Therapeutic Category No. Code Analgesic & Antipyretic / Muscle 1 1 Aceclofenac 100mg and Paracetamol 325 mg Tablet 10's 10's 8.00 relaxants Analgesic & Antipyretic / Muscle 2 2 Aceclofenac Tablets IP 100mg 10's 10's 4.37 relaxants Acetaminophen 325 + Tramadol Hydrochloride 37.5 film Analgesic & Antipyretic / Muscle 3 4 10's 8.00 coated Tablet 10's relaxants Analgesic & Antipyretic / Muscle 4 5 ASPIRIN Tablets IP 150 mg 14's 14's 2.70 relaxants DICLOFENAC 50 mg+ PARACETAMOL 325 mg+ Analgesic & Antipyretic / Muscle 5 6 10's 11.30 CHLORZOXAZONE 500 mg Tablets 10's relaxants Diclofenac Sodium 50mg + Serratiopeptidase 10mg Tablet Analgesic & Antipyretic / Muscle 6 8 10's 12.00 10's relaxants Analgesic & Antipyretic / Muscle 7 9 Diclofenac Sodium (SR) 100 mg Tablet 10's 10's 6.12 relaxants Analgesic & Antipyretic / Muscle 8 10 Diclofenac Sodium 25mg per ml Inj. IP 3 ml 3 ml 2.00 relaxants Analgesic & Antipyretic / Muscle 9 11 Diclofenac Sodium 50 mg Tablet 10's 10's 2.90 relaxants Analgesic & Antipyretic / Muscle 10 12 Etoricoxilb Tablets IP 120mg 10's 10's 33.00 relaxants Analgesic & Antipyretic / Muscle 11 13 Etoricoxilb Tablets IP 90mg 10's 10's 25.00 relaxants Analgesic & Antipyretic / Muscle 12 14 Ibuprofen 400 mg + Paracetamol 325 mg Tablet 10's 15's 5.50 relaxants Analgesic & Antipyretic / Muscle 13 15 Ibuprofen 200 mg film coated Tablet 10's 10's 1.80 relaxants Analgesic & Antipyretic / Muscle 14 16 Ibuprofen 400 mg film coated Tablet 10's 15's 3.50 relaxants Analgesic & Antipyretic -

Novel Derivatives of Bio-Affecting Phenolic Compounds and Pharmaceutical Composition Containing Them
Europaisches Patentamt European Patent Office © Publication number: 0046 270 A1 Office europeen des brevets ™ EUROPEAN PATENT APPLICATION @ Application number: 81106277.7 © Int. CI.3: C 07 C 103/78, C 07 C 93/26, C 07 C 69/24, C 07 C 1 53/07, @ Date of filing: 12.08.81 C07C 69/28 // C07C1 25/065 <§) Priority: 13.08.80 US 177825 © Applicant: INTERx RESEARCH CORPORATION, 2201 West 21 st Street, Lawrence Kansas 66044 (US) © I nventor : Bodor, Nicholas S., 31 5 Southwest 91 st Street, ® Dateofpublicationofapplication:24.02.82 S^^S^mHariMBM Bulletin m/b Terrace, Gainesville, Florida 32605 (US) Inventor: Pogany, Stefano A., 520 Louisiana Street, Lawrence Kansas 66044 (US) @ Designated Contracting States : AT BE CH DE FR GB IT ® Representative: Abitz, Walter, Dr.-lng. et al, Abitz, Mori, LI LU NL SE Gritschneder P.O. Box 86 01 09, D-8000 Munchen 86 (DE) Novel derivatives of bio-affecting phenolic compounds and pharmaceutical composition containing them. Novel@ Novel transient prodrug forms of bio-affecting phe- amyl, CH2ONO2,CH2ON02, -CH2OCOR2 or any non-heterocyclic nolic compounds are selected from the group consisting of member of the group defined by R2Rz above; and n.isn is at least those having the structural formula (I): one and equals the total number of phenolic hydroxyl functions comprising the non-steroidal bioaffecting phenol o etherified via a R2COXCH(R3)0-moiety; those having the structural formula (II): R2-C-X-CH-0- (I) I O R, II R2-C-X-CH-0- -RM-i-O-C-R2 (II) wherein X is O, S or NR5 wherein R5 is hydrogen or lower alkyl;alky!; -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

PHARMACEUTICAL APPENDIX to the TARIFF SCHEDULE 2 Table 1
Harmonized Tariff Schedule of the United States (2020) Revision 19 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2020) Revision 19 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names INN which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service CAS registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. -
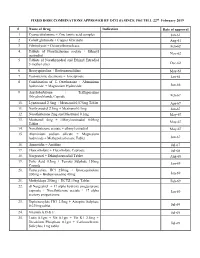
FIXED DOSE COMBINATIONS APPROVED by DCG (I) SINCE 1961 TILL 22Nd February 2019
FIXED DOSE COMBINATIONS APPROVED BY DCG (I) SINCE 1961 TILL 22nd February 2019 # Name of Drug Indication Date of approval 1. Cyanocobalamine + Zinc tannic acid complex Jan-61 2. Cobalt glutamate + Copper Glycinate Aug-61 3. Fibrinolysin + Desoxyribonuclease Feb-62 4. Tablets of Norethisterone acetate + Ethinyl Nov-62 oestradiol 5. Tablets of Norethynodrel and Ethinyl Estradiol 3-methyl ether Dec-62 6. Broxyquinoline + Brobenzoxalidine May-63 7. Testosterone decanoate + Isocaproate Jan-64 8. Combination of L Oxethazaine + Aluminium hydroxide + Magnesium Hydroxide Jun-66 9. Amylobarbitone + Trifluperazine Dihydrochloride Capsule Feb-67 10. Lynestronol 2.5mg + Mestranol 0.075mg Tablet Apr-67 11. Northynodrel 2.5mg + Mestranol 0.1mg Jun-67 12. Norethisterone 2mg and Mestranol 0.1mg May-67 13. Mestranol 4mg + Ethinyloestradial 0.05mg May-67 Tablet 14. Norethisterane acetate + ethinyl estradiol May-67 15. Aluminium sodium silicate + Magnesium hydroxide + Methypolysiloxane Tablet Jun-67 16. Ammoidin + Amidine Jul-67 17. Fluocortolene + Flucortolene Caproate Jul-68 18. Norgestrel + Ethinyloestradiol Tablet Aug-68 19. Folic Acid 0.5mg + Ferrous Sulphate 150mg Jan-69 Capsule 20. Tetracycline HCl 250mg + Broxyquinoline 200mg + Brobenzoxadine 40mg Jan-69 21. Methyldopa 250mg + HCTZ 15mg Tablet Feb-69 22. dl Norgestrel + 17 alpha hydroxy progesterone caproate + Norethisterone acetate + 17 alpha Jan-69 acetoxy progesterone 23. Diphenoxylate HCl 2.5mg + Atropine Sulphate 0.025mg tablet Jul-69 24. Vitamin A,D & E Jul-69 25. Lutin 0.1gm + Vit 0.1gm + Vit K1 2.5mg + Dicalcium Phosphate 0.1gm + Carlozochrome Jul-69 Salicylate 1mg tablet 26. Vit K 1 5mg + Calcium Lactolionate 100 m g+ Carlozocrome Salicylate 2.5mg + Phenol 0.5% Jul-69 + Lignocaine Hcl 1% injection 27. -

Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Cr
Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Crizotinib (PF-02341066) 1 4 55 Docetaxel 1 5 98 Anastrozole 1 6 25 Cladribine 1 7 23 Methotrexate 1 8 -187 Letrozole 1 9 65 Entecavir Hydrate 1 10 48 Roxadustat (FG-4592) 1 11 19 Imatinib Mesylate (STI571) 1 12 0 Sunitinib Malate 1 13 34 Vismodegib (GDC-0449) 1 14 64 Paclitaxel 1 15 89 Aprepitant 1 16 94 Decitabine 1 17 -79 Bendamustine HCl 1 18 19 Temozolomide 1 19 -111 Nepafenac 1 20 24 Nintedanib (BIBF 1120) 1 21 -43 Lapatinib (GW-572016) Ditosylate 1 22 88 Temsirolimus (CCI-779, NSC 683864) 1 23 96 Belinostat (PXD101) 1 24 46 Capecitabine 1 25 19 Bicalutamide 1 26 83 Dutasteride 1 27 68 Epirubicin HCl 1 28 -59 Tamoxifen 1 29 30 Rufinamide 1 30 96 Afatinib (BIBW2992) 1 31 -54 Lenalidomide (CC-5013) 1 32 19 Vorinostat (SAHA, MK0683) 1 33 38 Rucaparib (AG-014699,PF-01367338) phosphate1 34 14 Lenvatinib (E7080) 1 35 80 Fulvestrant 1 36 76 Melatonin 1 37 15 Etoposide 1 38 -69 Vincristine sulfate 1 39 61 Posaconazole 1 40 97 Bortezomib (PS-341) 1 41 71 Panobinostat (LBH589) 1 42 41 Entinostat (MS-275) 1 43 26 Cabozantinib (XL184, BMS-907351) 1 44 79 Valproic acid sodium salt (Sodium valproate) 1 45 7 Raltitrexed 1 46 39 Bisoprolol fumarate 1 47 -23 Raloxifene HCl 1 48 97 Agomelatine 1 49 35 Prasugrel 1 50 -24 Bosutinib (SKI-606) 1 51 85 Nilotinib (AMN-107) 1 52 99 Enzastaurin (LY317615) 1 53 -12 Everolimus (RAD001) 1 54 94 Regorafenib (BAY 73-4506) 1 55 24 Thalidomide 1 56 40 Tivozanib (AV-951) 1 57 86 Fludarabine -

(12) United States Patent (10) Patent No.: US 9,101,662 B2 Tamarkin Et Al
USOO91 01662B2 (12) United States Patent (10) Patent No.: US 9,101,662 B2 Tamarkin et al. (45) Date of Patent: *Aug. 11, 2015 (54) COMPOSITIONS WITH MODULATING A61K 47/32 (2013.01); A61 K9/0014 (2013.01); AGENTS A61 K9/0031 (2013.01); A61 K9/0034 (2013.01); A61 K9/0043 (2013.01); A61 K (71) Applicant: Foamix Pharmaceuticals Ltd., Rehovot 9/0046 (2013.01); A61 K9/0048 (2013.01); (IL) A61 K9/0056 (2013.01) (72) Inventors: Dov Tamarkin, Macabim (IL); Meir (58) Field of Classification Search Eini, Ness Ziona (IL); Doron Friedman, CPC ........................................................ A61 K9/12 Karmei Yosef (IL); Tal Berman, Rishon See application file for complete search history. le Ziyyon (IL); David Schuz, Gimzu (IL) (56) References Cited (73) Assignee: Foamix Pharmaceuticals Ltd., Rehovot U.S. PATENT DOCUMENTS (IL) 1,159,250 A 11/1915 Moulton (*) Notice: Subject to any disclaimer, the term of this 1,666,684 A 4, 1928 Carstens patent is extended or adjusted under 35 1924,972 A 8, 1933 Beckert 2,085,733. A T. 1937 Bird U.S.C. 154(b) by 0 days. 2,390,921 A 12, 1945 Clark This patent is Subject to a terminal dis 2,524,590 A 10, 1950 Boe claimer. 2,586.287 A 2/1952 Apperson 2,617,754 A 1 1/1952 Neely 2,767,712 A 10, 1956 Waterman (21) Appl. No.: 14/045,528 2.968,628 A 1/1961 Reed 3,004,894 A 10/1961 Johnson et al. (22) Filed: Oct. 3, 2013 3,062,715 A 11/1962 Reese et al. -

Electron Transfer Partners of Cytochrome P450
4 Electron Transfer Partners of Cytochrome P450 Mark J.l. Paine, Nigel S. Scrutton, Andrew W. Munro, Aldo Gutierrez, Gordon C.K. Roberts, and C. Roland Wolf 1. Introduction Although P450 redox partners are usually expressed independently, "self-sufficient" P450 monooxygenase systems have also evolved through Cytochromes P450 contain a heme center the fusion of P450 and CPR genes. These fusion where the activation of molecular oxygen occurs, molecules are found in bacteria and fungi, the best- resulting in the insertion of a single atom of known example being P450 BM3, a fatty acid oxygen into an organic substrate with the con (0-2 hydroxylase from Bacillus megaterium, which comitant reduction of the other atom to water. The comprises a soluble P450 with a fiised carboxyl- monooxygenation reaction requires a coupled and terminal CPR module (recently reviewed by stepwise supply of electrons, which are derived Munro^). BM3 has the highest catalytic activity from NAD(P)H and supplied via a redox partner. known for a P450 monooxygenase^ and was for P450s are generally divided into two major classes many years the only naturally occurring ftised sys (Class I and Class II) according to the different tem known until the identification of a eukaryotic types of electron transfer systems they use. P450s membrane-bound equivalent fatty acid hydroxy in the Class I family include bacterial and mito lase, CYP505A1, from the phytopathogenic fungus chondrial P450s, which use a two-component Fusarium oxysporurrP. A number of novel P450 sys shuttle system consisting of an iron-sulfur protein tems are starting to emerge from the large numbers (ferredoxin) and ferredoxin reductase (Figure 4.1). -

UC Berkeley UC Berkeley Electronic Theses and Dissertations
UC Berkeley UC Berkeley Electronic Theses and Dissertations Title Oriented Attachment of Cytochrome P450 2C9 to a Self-Assembled Monolayer on a Gold Electrode as a Biosensor Design Permalink https://escholarship.org/uc/item/1m67k8mm Author Schneider, Elizabeth Publication Date 2011 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California Oriented Attachment of Cytochrome P450 2C9 to a Self-Assembled Monolayer on a Gold Electrode as a Biosensor Design by Elizabeth Ann Schneider A dissertation submitted in partial satisfaction of the requirements for the degree of Joint Doctor of Philosophy with the University of California, San Francisco in Bioengineering in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Douglas S. Clark, Chair Associate Professor Shuvo Roy Professor Liwei Lin Dr. Robert Kostecki Fall 2011 Abstract Oriented Attachment of Cytochrome P450 2C9 to a Self-Assembled Monolayer on a Gold Electrode as a Biosensor Design by Elizabeth Ann Schneider Doctor of Philosophy in Bioengineering University of California, Berkeley Professor Douglas S. Clark, Chair Cytochrome P450s (CYPs) are a family of enzymes implicated in the metabolism of drugs in the body. Consequently, P450 reactions are of high interest to the pharmaceutical industry, where lead compounds in drug development are screened as potential substrates of CYPs. The P450 reaction involves electron transfer to an iron heme via NADPH and the electron transfer partner enzyme P450 reductase (CPR). By immobilizing CYPs on an electrode however, NADPH and CPR are potentially no longer needed and the immobilized CYP can act as a biosensor by accepting electrons directly from the electrode.