Molecular Neurobiological Clues to the Pathogenesis of Bipolar Disorder
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Landscape Genomic Approach to Investigate Genetic Adaptation in South African Indigenous Goat Populations by Khanyisile Mdladla
Landscape genomic approach to investigate genetic adaptation in South African indigenous goat populations by Khanyisile Mdladla Submitted in fulfilment of the academic requirements of Doctor of Philosophy in Genetics School of Life Sciences College of Agriculture, Engineering and Science University of KwaZulu-Natal Pietermaritzburg South Africa December 2016 PREFACE The research contained in this thesis was completed by the candidate while based in the Biotechnology Platform, Agricultural Research Council and the Discipline of Genetics, School of Life Sciences of the College of Agriculture, Engineering and Science, University of KwaZulu-Natal, Pietermaritzburg, South Africa. The research was financially supported by University of KwaZulu-Natal, National Research Foundation-Department of Science and Technology (NRF-DST) and the Agricultural Research Council. The contents of this work have not been submitted in any form to another university and, except where the work of others is acknowledged in the text, the results reported are due to investigations by the candidate. _________________________ Signed: Edgar Farai Dzomba Date: _________________________ Signed: Farai Catherine Muchadeyi Date: i DECLARATION 1: PLAGIARISM Note that two declaration sections are required if there are papers emanating from the dissertation/thesis. The first (obligatory) declaration concerns plagiarism and the second declaration specifies your role in the published papers. I, Khanyisile Mdladla declare that: (i) the research reported in this dissertation, except where otherwise indicated or acknowledged, is my original work; (ii) this dissertation has not been submitted in full or in part for any degree or examination to any other university; (iii) this dissertation does not contain other persons’ data, pictures, graphs or other information, unless specifically acknowledged as being sourced from other persons; (iv) this dissertation does not contain other persons’ writing, unless specifically acknowledged as being sourced from other researchers. -

Ce Document Est Le Fruit D'un Long Travail Approuvé Par Le Jury De Soutenance Et Mis À Disposition De L'ensemble De La Communauté Universitaire Élargie
AVERTISSEMENT Ce document est le fruit d'un long travail approuvé par le jury de soutenance et mis à disposition de l'ensemble de la communauté universitaire élargie. Il est soumis à la propriété intellectuelle de l'auteur. Ceci implique une obligation de citation et de référencement lors de l’utilisation de ce document. D'autre part, toute contrefaçon, plagiat, reproduction illicite encourt une poursuite pénale. Contact : [email protected] LIENS Code de la Propriété Intellectuelle. articles L 122. 4 Code de la Propriété Intellectuelle. articles L 335.2- L 335.10 http://www.cfcopies.com/V2/leg/leg_droi.php http://www.culture.gouv.fr/culture/infos-pratiques/droits/protection.htm Ecole Doctorale BioSE (Biologie-Santé-Environnement) Thèse Présentée et soutenue publiquement pour l’obtention du titre de DOCTEUR DE l’UNIVERSITE DE LORRAINE Mention : « Sciences de la Vie et de la Santé » Par Andréa GEOFFROY Conséquences d’une carence en donneurs de méthyles sur la différenciation neuronale et la plasticité : influence d’une supplémentation périnatale sur le développement cérébral. Le 08 Septembre 2015 Membres du jury : Rapporteurs : Pr. Marie-Laure Kottler PU-PH, U1075 INSERM, Caen, France Dr. Patrick Anglard DR, U7364 CNRS, Strasbourg, France Examinateurs : Dr. Hervé Moine DR, CNRS 7104 / INSERM U964, Strasbourg, France Pr. Jean-Louis Guéant PU-PH, U954 INSERM, Nancy, France Dr. Jean-Luc Daval DR, U954 INSERM, Nancy, France Directeur de thèse Dr. Carine Bossenmeyer-Pourié MCU, U954 INSERM, Nancy, France, Co-directeur de thèse Membres invités: Pr. Bruno Charpentier PR, U7365 CNRS, Nancy, France _________________________________________________________________________________ UMR 954 INSERM – «Nutrition, génétique et exposition aux risques environnementaux» 9 avenue de la Forêt de Haye-Faculté de Médecine - 54500 Vandoeuvre-lès-Nancy REMERCIEMENTS Je transmets mes sincères remerciements : A Madame le Professeur Marie-Laure Kottler et à Monsieur le Docteur Patrick Anglard pour avoir accepté d’être rapporteurs pour ma soutenance de thèse. -

SZDB: a Database for Schizophrenia Genetic Research
Schizophrenia Bulletin vol. 43 no. 2 pp. 459–471, 2017 doi:10.1093/schbul/sbw102 Advance Access publication July 22, 2016 SZDB: A Database for Schizophrenia Genetic Research Yong Wu1,2, Yong-Gang Yao1–4, and Xiong-Jian Luo*,1,2,4 1Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences and Yunnan Province, Kunming Institute of Zoology, Kunming, China; 2Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming, China; 3CAS Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences, Shanghai, China 4YGY and XJL are co-corresponding authors who jointly directed this work. *To whom correspondence should be addressed; Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China; tel: +86-871-68125413, fax: +86-871-68125413, e-mail: [email protected] Schizophrenia (SZ) is a debilitating brain disorder with a Introduction complex genetic architecture. Genetic studies, especially Schizophrenia (SZ) is a severe mental disorder charac- recent genome-wide association studies (GWAS), have terized by abnormal perceptions, incoherent or illogi- identified multiple variants (loci) conferring risk to SZ. cal thoughts, and disorganized speech and behavior. It However, how to efficiently extract meaningful biological affects approximately 0.5%–1% of the world populations1 information from bulk genetic findings of SZ remains a and is one of the leading causes of disability worldwide.2–4 major challenge. There is a pressing -

Identification and Characterisation of Rare CACNG5 Genetic Variants in Bipolar Disorder and Schizophrenia
Identification and characterisation of rare CACNG5 genetic variants in bipolar disorder and schizophrenia Thesis submitted for the degree of Doctor of Philosophy UCL Yi-Chun Lin Molecular Psychiatry Laboratory University College London 2010 – 2015 1 I, Yi-Chun Lin, confirm that all the work presented in this thesis is my own. I confirm that the information has been derived from other sources; it has been indicated in the thesis. 2 Acknowledgements This thesis would not have been completed without the help and support of colleagues and friends to whom I would now like to express my sincere thanks First and foremost of my thanks goes to my supervisor Dr Andrew McQuillin, who gave me this wonderful and exciting opportunity to carry out this PhD. His encouragement has provided tremendous support throughout my PhD. I am extremely grateful for his insightful advice, patience and kindness. I would also like to express my appreciation and thanks to Professor Hugh Gurling, my second supervisor, who provided further guidelines and support to enable me to complete this PhD research. Very special thanks to Dr. Radhika Kandaswamy and Dr. Sally Sharp, who have been guiding me with infinite patience in my experimental studies. I am indebted to them for their enthusiasm and advice, their friendship and support leading to the completion of research experiments. Also I would like to thank all my colleagues from my lab Michael, John, Alessia for their support. Finally, I would also like to thank my family, especially my parents for their financial support and my friends for their encouragement and support. -

RDH10 Is Necessary for Initial Vagal Neural Crest Cell Contribution to Embryonic Gastrointestinal Tract Development
RDH10 is Necessary for Initial Vagal Neural Crest Cell Contribution to Embryonic Gastrointestinal Tract Development BY © 2015 Naomi Elaine Butler Tjaden Submitted to the graduate degree program in Anatomy and Cell Biology and to the Graduate Faculty of The University of Kansas Medical Center in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Paul Trainor, Co-Chairperson Brenda Rongish, Co-Chairperson Timothy Fields Dale Abrahamson Osama Almadhoun Paul Cheney Date Defended: May 1, 2015 The Dissertation Committee for Naomi Elaine Butler Tjaden Certifies that this is the approved version of the following dissertation: RDH10 is Necessary for Initial Vagal Neural Crest Cell Contribution to Embryonic Gastrointestinal Tract Development Paul Trainor, Co-Chairperson Brenda Rongish, Co-Chairperson Date approved: May 5, 2015 ii Abstract Retinol dehydrogenase 10 (RDH10) catalyzes the first oxidative step in the metabolism of vitamin A to its active form retinoic acid (RA). Insufficient or excess RA can result in various congenital abnormalities, such as Hirschsprung disease (HSCR). In HSCR, neurons are absent from variable lengths of the gastrointestinal tract due to a failure of neural crest cell (NCC) colonization or development, leading to megacolon and/or the failure to pass meconium. Enteric neurons are derived from neural crest cells (NCC); hence HSCR is associated with incomplete NCC development or colonization of the GI tract. We investigated our hypothesis that RDH10 is necessary for proper NCC contribution to the enteric nervous system (ENS). The mouse point mutant Rdh10trex/trex exhibits decreased retinoid signaling and colonic aganglionosis. Organ explant culture and in utero retinal supplementation experiments define a temporal requirement for RA in ENS development between E7.5-E9.5. -

The Transcriptome of Utricle Hair Cell Regeneration in the Avian Inner Ear
The Journal of Neuroscience, March 5, 2014 • 34(10):3523–3535 • 3523 Development/Plasticity/Repair The Transcriptome of Utricle Hair Cell Regeneration in the Avian Inner Ear Yuan-Chieh Ku,1 Nicole A. Renaud,1 Rose A. Veile,1 Cynthia Helms,1 Courtney C.J. Voelker,2 Mark E. Warchol,2 and Michael Lovett1 1Department of Genetics and 2Department of Otolaryngology, Washington University School of Medicine, St Louis, Missouri 63110 Sensory hair cell loss is the major cause of hearing and balance disorders. Mammals are incapable of sustained hair cell regeneration, but lower vertebrates can regenerate these mechano-electrical transducers. We present the first comprehensive transcriptome (by mRNA- Seq) of hair cell regeneration in the chick utricle. We provide pathway and pattern annotations and correlate these with the phenotypic events that occur during regeneration. These patterns are surprisingly synchronous and highly punctuated. We show how these patterns are a new resource for identifying components of the hair cell transcriptome and identify 494 new putative hair-cell-specific genes and validate three of these (of three tested) by immunohistochemical staining. We describe many surprising new components and dynamic expression patterns, particularly within NOTCH signaling. For example, we show that HES7 is specifically expressed during utricle hair cell regeneration and closely parallels the expression of HES5. Likewise, the expression of ATOH1 is closely correlated with HEYL and the HLH inhibitory transcription factors ID1, ID2, and ID4. -

WO 2013/184908 A2 12 December 2013 (12.12.2013) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2013/184908 A2 12 December 2013 (12.12.2013) P O P C T (51) International Patent Classification: Jr.; One Procter & Gamble Plaza, Cincinnati, Ohio 45202 G06F 19/00 (201 1.01) (US). HOWARD, Brian, Wilson; One Procter & Gamble Plaza, Cincinnati, Ohio 45202 (US). (21) International Application Number: PCT/US20 13/044497 (74) Agents: GUFFEY, Timothy, B. et al; c/o The Procter & Gamble Company, Global Patent Services, 299 East 6th (22) Date: International Filing Street, Sycamore Building, 4th Floor, Cincinnati, Ohio 6 June 2013 (06.06.2013) 45202 (US). (25) Filing Language: English (81) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (30) Priority Data: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, 61/656,218 6 June 2012 (06.06.2012) US DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (71) Applicant: THE PROCTER & GAMBLE COMPANY HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KN, KP, KR, [US/US]; One Procter & Gamble Plaza, Cincinnati, Ohio KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 45202 (US). MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, (72) Inventors: XU, Jun; One Procter & Gamble Plaza, Cincin SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, nati, Ohio 45202 (US). -

1 SUPPLEMENTAL DATA Figure S1. Poly I:C Induces IFN-Β Expression
SUPPLEMENTAL DATA Figure S1. Poly I:C induces IFN-β expression and signaling. Fibroblasts were incubated in media with or without Poly I:C for 24 h. RNA was isolated and processed for microarray analysis. Genes showing >2-fold up- or down-regulation compared to control fibroblasts were analyzed using Ingenuity Pathway Analysis Software (Red color, up-regulation; Green color, down-regulation). The transcripts with known gene identifiers (HUGO gene symbols) were entered into the Ingenuity Pathways Knowledge Base IPA 4.0. Each gene identifier mapped in the Ingenuity Pathways Knowledge Base was termed as a focus gene, which was overlaid into a global molecular network established from the information in the Ingenuity Pathways Knowledge Base. Each network contained a maximum of 35 focus genes. 1 Figure S2. The overlap of genes regulated by Poly I:C and by IFN. Bioinformatics analysis was conducted to generate a list of 2003 genes showing >2 fold up or down- regulation in fibroblasts treated with Poly I:C for 24 h. The overlap of this gene set with the 117 skin gene IFN Core Signature comprised of datasets of skin cells stimulated by IFN (Wong et al, 2012) was generated using Microsoft Excel. 2 Symbol Description polyIC 24h IFN 24h CXCL10 chemokine (C-X-C motif) ligand 10 129 7.14 CCL5 chemokine (C-C motif) ligand 5 118 1.12 CCL5 chemokine (C-C motif) ligand 5 115 1.01 OASL 2'-5'-oligoadenylate synthetase-like 83.3 9.52 CCL8 chemokine (C-C motif) ligand 8 78.5 3.25 IDO1 indoleamine 2,3-dioxygenase 1 76.3 3.5 IFI27 interferon, alpha-inducible -

Antounians Lina 201511 Msc
An Inter-Vascular Bed and Inter-Species Investigation of Epigenetic Regulatory Elements in Endothelial Cells by Lina Antounians A thesis submitted in conformity with the requirements for the degree of Master’s of Science Department of Molecular Genetics University of Toronto © Copyright by Lina Antounians 2015 An Inter-Vascular Bed and Inter-Species Investigation of Epigenetic Regulatory Elements in Endothelial Cells Lina Antounians Master’s of Science Molecular Genetics, University of Toronto, 2015 Abstract A major challenge in human genetics is to understand the mechanisms that control gene expression. To identify gene regulatory regions required for vascular homeostasis, we performed chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) for a variety of histone modifications and the JUN transcription factor in primary aortic endothelial cells (ECs) isolated from human, rat (Rattus norvegicus) and bovine (Bos taurus). We generated a chromatin state map for human aortic ECs and found that the vast majority of regulatory regions in aortic ECs were also active in venous ECs. By comparing the genomic occupancy of JUN and a histone modification indicative of active enhancers (H3K27ac) between species, we identified a set of conserved regulatory regions that were enriched for EC-specific pathways and human regulatory disease variants. Overall, we demonstrate that comparative epigenomics is a viable strategy to identify functionally important vascular gene regulatory elements. ii Acknowledgements I would like to express my sincere gratitude to my supervisor Dr. Michael Wilson for his mentorship and encouragement throughout my Master’s degree. I have learned many technical and analytical skills in your lab, but none as important as the passion for learning, teaching, and collaborating with my peers. -

Table S1 Changes in the Expression Levels of Genes Induced by Sevoflurane
Table S1 Changes in the expression levels of genes induced by sevoflurane * Gene ID Gene abbreviation log2 ratio Rtn4rl2 Reticulon 4 receptor-like 2 7.80 Mas1 MAS1 oncogene 7.76 Gpx6 Glutathione peroxidase 6 7.62 Ovol2 Ovo-like 2 (Drosophila) 7.19 Calcr Calcitonin receptor 6.81 Impg1 Interphotoreceptor matrix proteoglycan 1 6.72 Neurod6 Neurogenic differentiation 6 6.33 Neurod2 Neurogenic differentiation 2 6.30 Dio3 Deiodinase, iodothyronine type III 5.91 Cd6 CD6 antigen 5.80 Gucy2g Guanylate cyclase 2g 5.75 Jsrp1 Junctional sarcoplasmic reticulum protein 1 5.56 Slc38a4 Solute carrier family 38, member 4 5.51 Tmprss6 Transmembrane serine protease 6 5.47 Sh3rf2 SH3 domain containing ring finger 2 5.41 Ccbp2 Chemokine binding protein 2 5.33 Gprc5a G protein-coupled receptor, family C, group 5, member A 5.27 Clspn Claspin homolog (Xenopus laevis) 5.21 Robo3 Roundabout homolog 3 (Drosophila) 5.21 Figf C-fos induced growth factor 5.20 Trpc6 Transient receptor potential cation channel, subfamily C, member 6 5.19 Adora2a Adenosine A2a receptor 5.12 Tbx15 T-box 15 5.07 Cdsn Corneodesmosin 5.02 Cd4 CD4 antigen 5.02 Col19a1 Collagen, type XIX, alpha 1 4.98 Kcnh3 Potassium voltage-gated channel, subfamily H (eag-related), member 3 4.94 Gpr88 G-protein coupled receptor 88 4.90 C1qtnf7 C1q and tumor necrosis factor related protein 7 4.88 Serine (or cysteine) peptidase inhibitor, clade A (alpha-1 antiproteinase, Serpina9 antitrypsin), member 9 4.87 Il20ra Interleukin 20 receptor, alpha 4.79 Lrg1 Leucine-rich alpha-2-glycoprotein 1 4.78 Rpe65 Retinal -

Characterization of Two G-Protein Coupled Receptors and One Fox Transcription Factor in Drosophila Embryonic Development by Cait
Characterization of Two G-Protein Coupled Receptors and One Fox Transcription Factor in Drosophila Embryonic Development By Caitlin D. Hanlon A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July 2015 ABSTRACT Cell migration is an exquisitely intricate process common to many higher organisms. Variations in the signals driving cell movement, the distance cells travel, and whether cells migrate as individuals, clusters, or as intact epithelia are all possible. Cell migration can be beneficial, as in development or wound healing, or detrimental, as in cancer metastasis. To begin to unravel the complexities inherent to cell migration, the Andrew lab uses the Drosophila salivary gland as a relatively simple model system for learning the molecular/cellular events underlying cell movement. The salivary gland begins as a placode of polarized columnar epithelial cells on the surface of the embryo that invaginates and move dorsally until a turning point is reached. There, it reorients and begins posterior migration, which continues until the gland reaches its final position along the anterior-posterior axis of the embryo. The broad goal of my work is to identify and characterize other key players in salivary gland migration. I characterized two G-protein coupled receptors (GPCRs) – Tre1 and mthl5 – which are expressed dynamically in the embryo. By creating a null allele of Tre1, I found that Tre1 plays a key role in germ cell migration and affects microtubule organization in the migrating salivary gland. I created a mthl5 mutant allele using the CRISPR/Cas9 system. mthl5 plays a role in the cell shape changes that drive salivary gland invagination. -
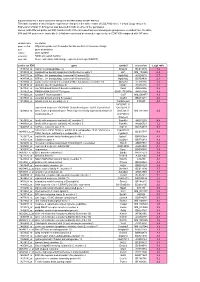
Supplementary File 1
Supplementary file 1: Gene expression changes in the white matter of CD47 KO mice This table contains a list of all gene expression changes in the white matter of CD47 KO mice ( > 2-fold: |Log2 ratio| > 1) Expression of total 14,875 genes was detected in both or either of the genotypes. Genes (with different probe set ID#) found in both of the increased and decreased gene groups were excluded from the table. 594 and 548 genes were markedly (> 2-fold) increased and decreased, respectively, in CD47 KO compared with WT mice. column name description probe set ID# Affymetrix probe set ID number for Mouse 430 2.0 Genome Arrays gene gene description symbol gene symbol accession NCBI accession number Log2 ratio Gene expression fold change expressed as Log2 (KO/WT) probe set ID# gene symbol accession Log2 ratio 1449153_at matrix metallopeptidase 12 Mmp12 BC019135 7.2 1419534_at oxidized low density lipoprotein (lectin-like) receptor 1 Olr1 NM_138648 6.2 1444176_at ATPase, H+ transporting, lysosomal V0 subunit D2 Atp6v0d2 AV204216 5.7 1434798_at ATPase, H+ transporting, lysosomal V0 subunit D2 Atp6v0d2 BB769890 2.3 1420504_at solute carrier family 6 (neurotransmitter transporter), member 14 Slc6a14 AF320226 5.5 1459934_at ubiquitin specific peptidase 42 Usp42 AA516835 5.2 1431800_at von Willebrand factor A domain containing 8 Vwa8 AK004956 5.2 1431927_at RIKEN cDNA 5033417F24 gene 5033417F24Rik AK018199 4.9 1419202_at cystatin F (leukocystatin) Cst7 NM_009977 4.9 1439943_at vacuolar protein sorting 54 (yeast) Vps54 BB201271 4.6 1419966_at tubulin,