Lipid Diene Conjugation in Human Saliva
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1D Atmospheric Study of the Temperate Sub-Neptune K2-18B D
A&A 646, A15 (2021) Astronomy https://doi.org/10.1051/0004-6361/202039072 & © D. Blain et al. 2021 Astrophysics 1D atmospheric study of the temperate sub-Neptune K2-18b D. Blain, B. Charnay, and B. Bézard LESIA, Observatoire de Paris, PSL Research University, CNRS, Sorbonne Université, Université de Paris, 92195 Meudon, France e-mail: [email protected] Received 30 July 2020 / Accepted 13 November 2020 ABSTRACT Context. The atmospheric composition of exoplanets with masses between 2 and 10 M is poorly understood. In that regard, the sub-Neptune K2-18b, which is subject to Earth-like stellar irradiation, offers a valuable opportunity⊕ for the characterisation of such atmospheres. Previous analyses of its transmission spectrum from the Kepler, Hubble (HST), and Spitzer space telescopes data using both retrieval algorithms and forward-modelling suggest the presence of H2O and an H2–He atmosphere, but have not detected other gases, such as CH4. Aims. We present simulations of the atmosphere of K2-18 b using Exo-REM, our self-consistent 1D radiative-equilibrium model, using a large grid of atmospheric parameters to infer constraints on its chemical composition. Methods. We compared the transmission spectra computed by our model with the above-mentioned data (0.4–5 µm), assuming an H2–He dominated atmosphere. We investigated the effects of irradiation, eddy diffusion coefficient, internal temperature, clouds, C/O ratio, and metallicity on the atmospheric structure and transit spectrum. Results. We show that our simulations favour atmospheric metallicities between 40 and 500 times solar and indicate, in some cases, the formation of H2O-ice clouds, but not liquid H2O clouds. -

Planet Hunters. VI: an Independent Characterization of KOI-351 and Several Long Period Planet Candidates from the Kepler Archival Data
Accepted to AJ Planet Hunters VI: An Independent Characterization of KOI-351 and Several Long Period Planet Candidates from the Kepler Archival Data1 Joseph R. Schmitt2, Ji Wang2, Debra A. Fischer2, Kian J. Jek7, John C. Moriarty2, Tabetha S. Boyajian2, Megan E. Schwamb3, Chris Lintott4;5, Stuart Lynn5, Arfon M. Smith5, Michael Parrish5, Kevin Schawinski6, Robert Simpson4, Daryll LaCourse7, Mark R. Omohundro7, Troy Winarski7, Samuel Jon Goodman7, Tony Jebson7, Hans Martin Schwengeler7, David A. Paterson7, Johann Sejpka7, Ivan Terentev7, Tom Jacobs7, Nawar Alsaadi7, Robert C. Bailey7, Tony Ginman7, Pete Granado7, Kristoffer Vonstad Guttormsen7, Franco Mallia7, Alfred L. Papillon7, Franco Rossi7, and Miguel Socolovsky7 [email protected] ABSTRACT We report the discovery of 14 new transiting planet candidates in the Kepler field from the Planet Hunters citizen science program. None of these candidates overlapped with Kepler Objects of Interest (KOIs) at the time of submission. We report the discovery of one more addition to the six planet candidate system around KOI-351, making it the only seven planet candidate system from Kepler. Additionally, KOI-351 bears some resemblance to our own solar system, with the inner five planets ranging from Earth to mini-Neptune radii and the outer planets being gas giants; however, this system is very compact, with all seven planet candidates orbiting . 1 AU from their host star. A Hill stability test and an orbital integration of the system shows that the system is stable. Furthermore, we significantly add to the population of long period 1This publication has been made possible through the work of more than 280,000 volunteers in the Planet Hunters project, whose contributions are individually acknowledged at http://www.planethunters.org/authors. -

Sirius Astronomer
FEBRUARY 2013 Free to members, subscriptions $12 for 12 Volume 40, Number 2 Jupiter is featured often in this newsletter because it is one of the most appealing objects in the night sky even for very small telescopes. The king of Solar System planets is prominent in the night sky throughout the month, located near Aldebaran in the constellation Taurus. Pat Knoll took this image on January 18th from his observing site at Kearney Mesa, California, using a Meade LX200 Classic at f/40 with a 4X Powermate. The image was compiled from a two-minute run with an Imaging Source DFK 21AU618.AS camera . OCA CLUB MEETING STAR PARTIES COMING UP The free and open club meeting The Black Star Canyon site will open The next session of the Beginners will be held February 8 at 7:30 PM on February 2. The Anza site will be Class will be held at the Heritage in the Irvine Lecture Hall of the open on February 9. Members are en- Museum of Orange County at Hashinger Science Center at couraged to check the website calen- 3101 West Harvard Street in San- Chapman University in Orange. dar for the latest updates on star par- ta Ana on February 1st. The fol- This month, UCSD’s Dr. Burgasser ties and other events. lowing class will be held March will present “The Coldest Stars: Y- 1st Dwarfs and the Fuzzy Border be- Please check the website calendar for tween Stars and Planets.” the outreach events this month! Volun- GOTO SIG: TBA teers are always welcome! Astro-Imagers SIG: Feb. -

Antanamo Bay 'Ocr'' Er T "
1 rC'y!0'% %04 -0 Guantanamo Bay 'ocr'' er t ". ~ O " ~al kr "" Vol. 58 No. 3 Friday, January 19, 2001 Base residents take part Lassiter, Kochan selected in Candlelight March Naval Base SOY, JSOY GUANTANAMO BAY - EM1(SW) Wilbur A. Lasseter from the U.S. Naval Brig was recently selected as 2000 Sailor of the Year for Naval Station and Naval Base. He was also selected as Sailor of the Quarter for the fourth quarter for Naval Station and Naval Base. Lasseter will now compete for Southeast Region Sailor of the Year in Jacksonville the week of Feb. 11. Lasseter served as brig guard and recently elevated to brig administra- tive supervisor because of his un- matched leadership and managerial a skills. Through his superb leadership and hard work, Lasseter achieved a Com- bined Federal Campaign contribution for the Naval Station in excess of $21,000, 55 percent of the Naval +. Base's total goal of $38,000. As a brig career counselor, his efforts yielded three reenlistments, three Overseas Tour Extension Incen- tive Program requests and one con- version package. In addition, Lasseter performed a host of formal and HMI Tim Hill delivers a "moving" version of Dr Martin Luther informal counseling sessions. King famous "IHave A Dream" speech at POW/MIA Memorial - continued on page two (top). More than 320 GTMO residents marched in the 2nd annual CandlelightMarch in honor ofDr Martin Luther King Jr. GUANTANAMO BAY - BU3 Joseph J. Kochan was recently se- Photos by JOC Walter T Ham IV (top) lected as the Naval Station and Naval Base 2000 Junior Sailor of the and PH2 Emnitt J Hawks (bottom) Year and the 4th quarter Sailor of the Quarter. -

Basin Name 8-Digit HUC/ Service Area Owner Type Project Name County Corps AID No
updated February 28, 2021 Mitigation Requirements DWR Permit Warm Cool Cold Unspecified Riparian Non- Coastal Riparian Basin Name 8-digit HUC/ Service Area Owner Type Project Name County Corps AID No. Payment Date Payment amount Stream Stream Stream Thermal Wetland Riparian Wetland Buffer (sqft) PASQUOTANK 03010205 Private Barrier Island Station DARE 1996-0710 6 /23/1997 $18,000.00 0 0 0 0.00 1.44 0.00 0.00 CAPE FEAR 03030002 Private Panther Creek WAKE 1996-0320 8 /10/1997 $18,000.00 0 0 0 0.61 0.00 0.00 0.00 CAPE FEAR 03030003 Private Bailey Farms WAKE 1995-0102 9 /3 /1997 $12,000.00 0 0 0 0.40 0.00 0.00 0.00 FRENCH BROAD 06010105 DOT DOT - Widening of NC 151 BUNCOMBE 1997-0440 10/2 /1997 $32,500.00 0 0 0 260 0.00 0.00 0.00 0.00 CAPE FEAR 03030002 Private Kit Creek WAKE 1997-08149 1997-0729 10/14/1997 $6,000.00 0 0 0 0.25 0.00 0.00 0.00 FRENCH BROAD 06010105 Private Nasty Branch BUNCOMBE 1997-0123 11/13/1997 $37,500.00 0 0 0 300 0.00 0.00 0.00 0.00 CAPE FEAR 03030005 Private Motts Creek Apartments NEW HANOVER 1997-0075 12/11/1997 $44,280.00 0 0 0 0.00 3.69 0.00 0.00 NEUSE 03020201 Government Town of Cary - Cary Parkway Extension WAKE 1997-0227 1 /30/1998 $56,250.00 450 0 0 0 0.00 0.00 0.00 0.00 CAPE FEAR 03030002 Private Amberly Development CHATHAM 1995-0410 2 /2 /1998 $30,000.00 240 0 0 0 0.00 0.00 0.00 0.00 CATAWBA 03050103 Private McKee Road Apartments MECKLENBURG 1997-0809 2 /13/1998 $27,500.00 220 0 0 0 0.00 0.00 0.00 0.00 CAPE FEAR 03030002 Private Northeast Creek Parkway DURHAM 1997-0575 3 /19/1998 $22,750.00 182 0 0 0 0.00 0.00 -

Ceres: Astrobiological Target and Possible Ocean World
ASTROBIOLOGY Volume 20 Number 2, 2020 Research Article ª Mary Ann Liebert, Inc. DOI: 10.1089/ast.2018.1999 Ceres: Astrobiological Target and Possible Ocean World Julie C. Castillo-Rogez,1 Marc Neveu,2,3 Jennifer E.C. Scully,1 Christopher H. House,4 Lynnae C. Quick,2 Alexis Bouquet,5 Kelly Miller,6 Michael Bland,7 Maria Cristina De Sanctis,8 Anton Ermakov,1 Amanda R. Hendrix,9 Thomas H. Prettyman,9 Carol A. Raymond,1 Christopher T. Russell,10 Brent E. Sherwood,11 and Edward Young10 Abstract Ceres, the most water-rich body in the inner solar system after Earth, has recently been recognized to have astrobiological importance. Chemical and physical measurements obtained by the Dawn mission enabled the quantification of key parameters, which helped to constrain the habitability of the inner solar system’s only dwarf planet. The surface chemistry and internal structure of Ceres testify to a protracted history of reactions between liquid water, rock, and likely organic compounds. We review the clues on chemical composition, temperature, and prospects for long-term occurrence of liquid and chemical gradients. Comparisons with giant planet satellites indicate similarities both from a chemical evolution standpoint and in the physical mechanisms driving Ceres’ internal evolution. Key Words: Ceres—Ocean world—Astrobiology—Dawn mission. Astro- biology 20, xxx–xxx. 1. Introduction these bodies, that is, their potential to produce and maintain an environment favorable to life. The purpose of this article arge water-rich bodies, such as the icy moons, are is to assess Ceres’ habitability potential along the same lines Lbelieved to have hosted deep oceans for at least part of and use observational constraints returned by the Dawn their histories and possibly until present (e.g., Consolmagno mission and theoretical considerations. -

Exoplanet Secondary Atmosphere Loss and Revival 2 3 Edwin S
1 Exoplanet secondary atmosphere loss and revival 2 3 Edwin S. Kite1 & Megan Barnett1 4 1. University of Chicago, Chicago, IL ([email protected]). 5 6 Abstract. 7 8 The next step on the path toward another Earth is to find atmospheres similar to those of 9 Earth and Venus – high-molecular-weight (secondary) atmospheres – on rocky exoplanets. 10 Many rocky exoplanets are born with thick (> 10 kbar) H2-dominated atmospheres but 11 subsequently lose their H2; this process has no known Solar System analog. We study the 12 consequences of early loss of a thick H2 atmosphere for subsequent occurrence of a high- 13 molecular-weight atmosphere using a simple model of atmosphere evolution (including 14 atmosphere loss to space, magma ocean crystallization, and volcanic outgassing). We also 15 calculate atmosphere survival for rocky worlds that start with no H2. Our results imply that 16 most rocky exoplanets interior to the Habitable Zone that were formed with thick H2- 17 dominated atmospheres lack high-molecular-weight atmospheres today. During early 18 magma ocean crystallization, high-molecular-weight species usually do not form long-lived 19 high-molecular-weight atmospheres; instead they are lost to space alongside H2. This early 20 volatile depletion makes it more difficult for later volcanic outgassing to revive the 21 atmosphere. The transition from primary to secondary atmospheres on exoplanets is 22 difficult, especially on planets orbiting M-stars. However, atmospheres should persist on 23 worlds that start with abundant volatiles (waterworlds). Our results imply that in order to 24 find high-molecular-weight atmospheres on warm exoplanets orbiting M-stars, we should 25 target worlds that formed H2-poor, that have anomalously large radii, or which orbit less 26 active stars. -

Building an Energy Future
BUILDING AN ENERGY FUTURE INVESTORS’ HANDBOOK ROYAL DUTCH SHELL PLC FINANCIAL AND 12'4#6+10#.+0(14/#6+10s 2 36 61 COMPANY OVERVIEW PROJECTS & TECHNOLOGY UPSTREAM DATA 2 1WTDWUKPGUUGU 37 +PPQXCVKXGVGEJPQNQI[ 61 7RUVTGCOGCTPKPIU 4 *KIJNKIJVU 37 &GNKXGTKPIRTQLGEVU 63 1KNCPFICUGZRNQTCVKQPCPF 5 5VTCVGI[CPFQWVNQQM 38 %QPVTCEVKPICPFRTQEWTGOGPV RTQFWEVKQPCEVKXKVKGUGCTPKPIU 9 -G[RTQLGEVUWPFGTEQPUVTWEVKQP 38 5CHGV[ 65 1KNUCPFU 11 1RVKQPUHQTHWVWTGITQYVJ 38 4&GZRGPFKVWTG 66 2TQXGFQKNCPFICUTGUGTXGU 12 /CTMGVQXGTXKGY 69 1KNICUU[PVJGVKEETWFGQKNCPF 13 4GUWNVU DKVWOGPRTQFWEVKQP 72 #ETGCIGCPFYGNNU 39 74 .0)CPF)6. 14 CORPORATE SEGMENT UPSTREAM 75 15 'ZRNQTCVKQP 40 DOWNSTREAM DATA 18 2TQXGFTGUGTXGU MAPS 18 2TQFWEVKQP 75 1KNRTQFWEVUCPFTGƂPKPINQECVKQPU 19 +PVGITCVGFICU 40 'WTQRG 77 1KNUCNGUCPFTGVCKNUKVGU 21 9KPF 42 #HTKEC 78 %JGOKECNUCPFOCPWHCEVWTKPI 22 'WTQRG 44 #UKC NQECVKQPU 23 #HTKEC 48 1EGCPKC 24 #UKC KPENWFKPI/KFFNG'CUVCPF4WUUKC 49 #OGTKECU 27 1EGCPKC 28 #OGTKECU 80 ADDITIONAL INVESTOR 52 INFORMATION CONSOLIDATED DATA 30 80 5JCTGKPHQTOCVKQP DOWNSTREAM 52 'ORNQ[GGU 81 &KXKFGPFU 53 %QPUQNKFCVGFƂPCPEKCNFCVC 82 $QPFJQNFGTKPHQTOCVKQP 31 /CPWHCEVWTKPI 83 (KPCPEKCNECNGPFCT 32 5WRRN[CPFFKUVTKDWVKQP 32 $WUKPGUUVQ$WUKPGUU $$ 33 4GVCKN 33 .WDTKECPVU 33 .0)HQTVTCPURQTV 34 %JGOKECNU 34 6TCFKPI 34 $KQHWGNU 35 2QTVHQNKQCEVKQPU ABOUT THIS PUBLICATION 6JKU+PXGUVQTUo*CPFDQQMEQPVCKPUFGVCKNGFKPHQTOCVKQPCDQWV QWTCPPWCNƂPCPEKCNCPFQRGTCVKQPCNRGTHQTOCPEGQXGTXCT[KPI VKOGUECNGUHTQOVQ9JGTGXGTRQUUKDNGVJGHCEVU CPFƂIWTGUJCXGDGGPOCFGEQORCTCDNG6JGKPHQTOCVKQP KPVJKURWDNKECVKQPKUDGUVWPFGTUVQQFKPEQODKPCVKQPYKVJVJG -

Building Protoplanetary Disks from the Molecular Cloud: Redefining the Disk Timeline K
A&A 624, A93 (2019) Astronomy https://doi.org/10.1051/0004-6361/201730677 & © K. Baillié et al. 2019 Astrophysics Building protoplanetary disks from the molecular cloud: redefining the disk timeline K. Baillié1,2, J. Marques3, and L. Piau4 1 IMCCE, Observatoire de Paris, PSL Research University, CNRS, Sorbonne Universités, UPMC University Paris 06, Université Lille, 77 Av. Denfert-Rochereau, 75014 Paris, France e-mail: [email protected] 2 Centre National d’Études Spatiales, 2 place Maurice Quentin, 75039 Paris Cedex 01, France 3 Université Paris-Sud, Institut d’Astrophysique Spatiale, UMR 8617, CNRS, Batiment 121, 91405 Orsay Cedex, France 4 77 avenue Denfert-Rochereau, 75014 Paris, France Received 22 February 2017 / Accepted 26 February 2019 ABSTRACT Context. Planetary formation models are necessary to understand the characteristics of the planets that are the most likely to survive. Their dynamics, their composition and even the probability of their survival depend on the environment in which they form. We therefore investigate the most favorable locations for planetary embryos to accumulate in the protoplanetary disk: the planet traps. Aims. We study the formation of the protoplanetary disk by the collapse of a primordial molecular cloud, and how its evolution leads to the selection of specific types of planets. Methods. We use a hydrodynamical code that accounts for the dynamics, thermodynamics, geometry and composition of the disk to numerically model its evolution as it is fed by the infalling cloud material. As the mass accretion rate of the disk onto the star determines its growth, we can calculate the stellar characteristics by interpolating its radius, luminosity and temperature over the stellar mass from pre-calculated stellar evolution models. -
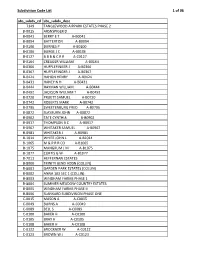
Subdivision Code List
Subdivision Code List 1 of 96 abs_subdv_cd abs_subdv_desc 7349 TANGLEWOOD AIRPARK ESTATES PHASE 2 B‐0015 ARSNSPIGER D B‐0041 BERRY E T A‐B0041 B‐0094 BATTERTON A‐B0094 B‐0100 BURNES P A‐B0100 B‐0106 BURGE J C A‐B0106 B‐0127 B B B & C R R A‐C0127 B‐0164 CREAGER WILLIAM A‐B0164 B‐0366 HUFFLEFINGER J A‐B0366 B‐0367 HUFFLEFINGER J A‐B0367 B‐0424 HAHON HENRY A‐B0424 B‐0431 HANEY N H A‐B0431 B‐0444 HAYMAN WILLIAM A‐B0444 B‐0492 JACKSON WILLIAM T A‐B0492 B‐0720 PRUETT SAMUEL A‐B0720 B‐0742 ROBERTS MARK A‐B0742 B‐0795 SWEETENBURG FRED A‐B0795 B‐0872 SLAYBURN JOHN A‐B0872 B‐0902 TATE CYNTHIA A‐B0902 B‐0917 THOMPSON B C A‐B0917 B‐0967 WHITAKER SAMUEL A‐B0967 B‐0981 WHITAKER J A‐B0981 B‐1014 WHITE JOHN L A‐B1014 B‐1065 M & P R R CO A‐B1065 B‐1075 MANGRUM J M A‐B1075 B‐1077 CURTIS G W A‐B1077 B‐7011 HEFFERNAN ESTATES B‐8000 TRINITY BEND ADDN (COLLIN) B‐8001 GARDEN PARK ESTATES (COLLIN) B‐8002 ANNA 103 SEC 1 (COLLIN) B‐8003 WINDHAM FARMS PHASE 1 B‐8004 SUMMER MEADOW COUNTRY ESTATES B‐8005 WINDHAM FARMS PHASE II B‐8006 SLANKARD SUBDIVISION PHASE ONE C‐0015 MASON A A‐C0015 C‐0049 BURNS A A‐C0049 C‐0089 BELL S A‐C0089 C‐0100 BAKER H A‐C0100 C‐0105 BRAY H A‐C0105 C‐0108 BAKER H A‐C0108 C‐0122 BROCKNER W A‐C0122 C‐0123 BROWN W J A‐C0123 Subdivision Code List 2 of 96 abs_subdv_cd abs_subdv_desc C‐0126 BRENHAM R F A‐C0126 C‐0226 CLARK J R A‐C0226 C‐0227 CLARK J R A‐C0227 C‐0261 CUMBERLAND G A‐C0261 C‐0281 CROMWELL & HAWLEY A‐C0281 C‐0300 DUGAN D A‐C0300 C‐0317 DYE ROMA A‐C0317 C‐0338 DUNCAN D A‐C0338 C‐0348 EDMISTON S A‐C0348 C‐0354 EDMISTON H A‐C0354 C‐0360 -

A Paucity of Proto-Hot Jupiters on Super-Eccentric Orbits
Submitted to ApJ on October 26th, 2012. Accepted on October 20th, 2014. Preprint typeset using LATEX style emulateapj v. 5/2/11 THE PHOTOECCENTRIC EFFECT AND PROTO-HOT JUPITERS III: A PAUCITY OF PROTO-HOT JUPITERS ON SUPER-ECCENTRIC ORBITS Rebekah I. Dawson1,2,3,7, Ruth A. Murray-Clay3,4, and John Asher Johnson3,5,6 Submitted to ApJ on October 26th, 2012. Accepted on October 20th, 2014. ABSTRACT Gas giant planets orbiting within 0.1 AU of their host stars, unlikely to have formed in situ, are evidence for planetary migration. It is debated whether the typical hot Jupiter smoothly migrated inward from its formation location through the proto-planetary disk or was perturbed by another body onto a highly eccentric orbit, which tidal dissipation subsequently shrank and circularized during close stellar passages. Socrates and collaborators predicted that the latter class of model should produce a population of super-eccentric proto-hot Jupiters readily observable by Kepler. We find a paucity of such planets in the Kepler sample, inconsistent with the theoretical prediction with 96.9% confidence. Observational effects are unlikely to explain this discrepancy. We find that the fraction of hot Jupiters with orbital period P > 3 days produced by the star-planet Kozai mechanism does not exceed (at two-sigma) 44%. Our results may indicate that disk migration is the dominant channel for producing hot Jupiters with P > 3 days. Alternatively, the typical hot Jupiter may have been perturbed to a high eccentricity by interactions with a planetary rather than stellar companion and began tidal circularization much interior to 1 AU after multiple scatterings. -
To View Poster
Planet Hunters Update: Many New Planet Candidates Identified by Citizen Scientists from Kepler Data, Including Several in the Habitable Zone Joseph R. Schmitt, Ji Wang, and the Planet Hunters team1 Abstract Since December, 2010, more than 250,000 public volunteers have searched through more than 19 million Kepler light curves hunting for transiting planets. The Kepler light curves are shown in 30 day sections, and with ~160,000 Kepler target stars, the users have contributed the equivalent of 180 years of work hours. This vetting process has resulted in over 40 new planet candidates and two new confirmed planets, including several not identified through the Kepler pipeline. Many of our candidate planets lie within their host star’s habitable zone. We review the recent large release of new PH candidates in Wang et al. (2013), including one confirmed planet, and give preliminary results for our next PH candidate release. The Planet Hunters Project The Planet Hunters project (www.planethunters.org), launched in December, 2010 as a member of the Zooniverse network, has citizen scientists visually examine Kepler archival data for potential planetary transit signals. While the Kepler’s Transit Planet Search (TPS) algorithm (Jenkins et al. 2002, 2010) has clearly been very successful, Planet Hunters have found a number of signals missed by the TPS algorithm. The process begins with Planet Hunters being shown a 30 day light curve. The user will mark the stellar variability and whether transit features are seen (see Figure 1). Planet Hunters are ≳ 85% efficient at finding planets above 4 R⊕, but are still capable of finding weaker signals (Schwamb et al.