SKIP Counteracts P53-Mediated Apoptosis Via Selective Regulation of P21cip1 Mrna Splicing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

In Silico Analysis of Regulatory Elements of the Vitamin D Receptor
Open Access Baghdad Science Journal P-ISSN: 2078-8665 2020, 17(2):463-470 E-ISSN: 2411-7986 DOI: http://dx.doi.org/10.21123/bsj.2020.17.2.0463 In Silico Analysis of Regulatory Elements of the Vitamin D Receptor Shirin Farivar 1 Roya Amirinejad 2 Bahar Naghavi gargari 3 Seyedeh Batool Hassani 4 Zeinab Shirvani-Farsani 1* Received 26/2/2019, Accepted 5/1/2020, Published 1/6/2020 This work is licensed under a Creative Commons Attribution 4.0 International License. Abstract Vitamin D receptor (VDR) is a nuclear transcription factor that controls gene expression. Its impaired expression was found to be related to different diseases. VDR also acts as a regulator of different pathways including differentiation, inflammation, calcium and phosphate absorption, etc. but there is no sufficient knowledge about the regulation of the gene itself. Therefore, a better understanding of the genetic and epigenetic factors regulating the VDR may facilitate the improvement of strategies for the prevention and treatment of diseases associated with dysregulation of VDR. In the present investigation, a set of databases and methods were used to identify putative functional elements in the VDR locus. Histone modifications, CpG Islands, epigenetic marks at VDR locus were indicated. In addition, repeated sequences, enhancers, insulators, transcription factor binding sites and targets of the VDR gene, as well as protein- protein interactions with bioinformatics tools, were reported. Some of these genetic elements had overlapped with CpG Islands. These results revealed important new insight into the molecular mechanisms of the VDR gene regulation in human cells and tissues. Key words: CpG Islands, Epigenetics, In silico analysis, Regulation elements, VDR Introduction: Vitamin D is a steroid hormone that multiple sclerosis (10), Rheumatoid Arthritis and balances calcium and phosphate levels and is Systemic Lupus Erythematosus (11), cardiovascular crucial for bone structure. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

The SMAD-Binding Domain of SKI: a Hotspot for De Novo Mutations Causing Shprintzen&Ndash
European Journal of Human Genetics (2015) 23, 224–228 & 2015 Macmillan Publishers Limited All rights reserved 1018-4813/15 www.nature.com/ejhg ARTICLE The SMAD-binding domain of SKI: a hotspot for de novo mutations causing Shprintzen–Goldberg syndrome Dorien Schepers1,20, Alexander J Doyle2,3,20,GretchenOswald2, Elizabeth Sparks2,LorethaMyers2,PatrickJWillems4, Sahar Mansour5, Michael A Simpson6,HelenaFrysira7, Anneke Maat-Kievit8,RickVanMinkelen8, Jeanette M Hoogeboom8, Geert R Mortier1, Hannah Titheradge9,LouiseBrueton9,LoisStarr10, Zornitza Stark11, Charlotte Ockeloen12, Charles Marques Lourenco13,EdBlair14, Emma Hobson15,JaneHurst16, Isabelle Maystadt17, Anne Destre´e17, Katta M Girisha18, Michelle Miller19,HarryCDietz2,3,BartLoeys1,20 and Lut Van Laer*,1,20 Shprintzen–Goldberg syndrome (SGS) is a rare, systemic connective tissue disorder characterized by craniofacial, skeletal, and cardiovascular manifestations that show a significant overlap with the features observed in the Marfan (MFS) and Loeys–Dietz syndrome (LDS). A distinguishing observation in SGS patients is the presence of intellectual disability, although not all patients in this series present this finding. Recently, SGS was shown to be due to mutations in the SKI gene, encoding the oncoprotein SKI, a repressor of TGFb activity. Here, we report eight recurrent and three novel SKI mutations in eleven SGS patients. All were heterozygous missense mutations located in the R-SMAD binding domain, except for one novel in-frame deletion affecting the DHD domain. Adding our new findings to the existing data clearly reveals a mutational hotspot, with 73% (24 out of 33) of the hitherto described unrelated patients having mutations in a stretch of five SKI residues (from p.(Ser31) to p.(Pro35)). -

Epitect® Methyl II Signature PCR Array (22) Human Notch Signaling Pathway
EpiTect® Methyl II Signature PCR Array (22) Human Notch Signaling Pathway Cat. no. 335212 EAHS-611ZA For DNA methylation analysis using MethylScreen™ technology The Human Notch Signaling Pathway EpiTect Methyl II Signature PCR Array profiles the promoter methylation status of a panel of 22 genes central to Notch signal transduction. The Notch signaling pathway regulates the expression of genes mediating cell-cell communication and developmental processes. DNA methylation status changes on key regulatory genes during development affect their ability to be induced or repressed by Notch and other signaling pathways. Profiling research samples containing genomic DNA from cellular or fresh tissue with this array may help to correlate CpG island methylation status with biological phenotypes such as developmental stages. This array contains key ligands, cofactors, receptors, downstream signaling effectors, and target genes for Notch signaling. The results may also help provide further insights into the molecular mechanisms and biological pathways behind the role of Notch signaling during cellular communication, differentiation, and development. With a simple restriction enzyme digestion and real-time PCR, research studies can analyze the promoter methylation status of 22 different genes central to Notch signal transduction with this DNA methylation PCR array. The EpiTect Methyl II PCR Arrays use MethylScreen™ technology provided under license from Orion Genomics, LLC. For further details, consult the EpiTect Methyl II PCR Array Handbook. Shipping and storage EpiTect Methyl II Signature PCR Arrays are shipped at room temperature (15–25°C), on dry ice, or on blue ice depending on the destination and accompanying products. They should be stored at –20°C. -
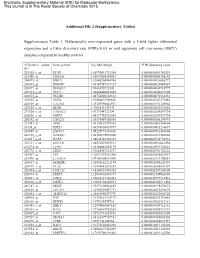
Differentially Over-Expressed Genes with a 2-Fold Higher Differe
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2015 Additional File 2 (Supplementary Tables) Supplementary Table 1: Differentially over-expressed genes with a 2-fold higher differential expression and a False discovery rate (FDR)<0.05 in oral squamous cell carcinoma (OSCC) samples compared to healthy controls. Affymetrix probe Gene symbol Log fold change FDR adjusted-p value ID 209365_s_at ECM1 1.65970411718384 0.0000000001342029 211980_at COL4A1 1.63093004254041 0.0000000003388142 204475_at MMP1 1.21640208064746 0.0000000016668772 206605_at ENDOU 1.49187987837927 0.0000000020488567 218677_at S100A14 1.5666356921542 0.0000000030718997 201325_s_at EMP1 1.42488420822805 0.0000000042431990 203675_at NUCB2 1.48754002522513 0.0000000076914233 218935_at EHD3 1.51884627509609 0.0000000147373416 220149_at C2orf54 1.35307996462937 0.0000000151328942 207655_s_at BLNK 1.4981534243447 0.0000000230316858 217508_s_at C18orf25 1.4737344722194 0.0000000259591734 203936_s_at MMP9 1.54117752982424 0.0000000259591734 205242_at CXCL13 1.36176459188585 0.0000000265290993 212543_at AIM1 1.43105615599363 0.0000000400764664 38158_at ESPL1 1.43594803693999 0.0000000415216687 203585_at ZNF185 1.54225773236818 0.0000000521483660 220330_s_at SAMSN1 1.48385675070206 0.0000000531560858 212657_s_at IL1RN 1.38628748328333 0.0000000754734933 202311_s_at COL1A1 1.44526073425523 0.0000000810641434 212274_at LPIN1 1.28166083845179 0.0000000951352623 209772_s_at CD24 1.39648587121677 0.0000000951352623 203407_at -

Anti-SNW1-Antibody Pack Size
TECHNICAL DATA SHEET Cat No: Kab08081 Product Particulars: anti-SNW1-antibody Pack Size: 100μg Pack Size: Pack Size: 100μg Purify: Immunogen affinity purified Host: Rabbit Isotype: IgG Storage: PBS with 0.02% sodium azide and 50% glycerol pH 7.3 , -20Ԩ for 24 months (Avoid repeated freeze / thaw cycles.) Background (Function): Involved in transcriptional regulation. Modulates TGF- beta-mediated transcription via association with SMAD proteins, MYOD1-mediated transcription via association with PABPN1, RB1- mediated transcriptional repression, and retinoid-X receptor (RXR)- and vitamin D receptor (VDR)-dependent gene transcription in a cell line-specific manner probably involving coactivators NCOA1 and GRIP1. Is involved in NOTCH1-mediated transcriptional activation. Binds to multimerized forms of Notch intracellular domain (NICD) and is proposed to recruit transcriptional coactivators such as MAML1 to form an intermediate preactivation complex which associates with DNA-bound CBF-1/RBPJ to form a transcriptional activation complex by releasing SNW1 and redundant NOTCH1 NICD. Proposed to be involved in transcriptional activation by EBV EBNA2 of CBF-1/RBPJ-repressed promoters. Is recruited by HIV-1 Tat to Tat:P-TEFb:TAR RNA complexes and is involved in Tat transcription by recruitment of MYC, MEN1 and TRRAP to the HIV promoter. Functions as a splicing factor in pre-mRNA splicing. Is required in the specific splicing of CDKN1A pre- mRNA; the function probbaly involves the recruitment of U2AF2 to the mRNA. Is proposed to recruit PPIL1 to the spliceosome. May be involved in cyclin-D1/CCND1 mRNA stability through the SNARP complex which associates with both the 3'end of the CCND1 gene and its mRNA. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

Genomic Alterations in High-Risk Chronic Lymphocytic Leukemia Frequently Affect Cell Ferrata Storti Foundation Cycle Key Regulators and NOTCH1-Regulated Transcription
Chronic Lymphocytic Leukemia ARTICLE Genomic alterations in high-risk chronic lymphocytic leukemia frequently affect cell Ferrata Storti Foundation cycle key regulators and NOTCH1-regulated transcription Jennifer Edelmann,1,2 Karlheinz Holzmann,3 Eugen Tausch,1 Emily A. Saunderson,2 Billy M. C. Jebaraj,1 Daniela Steinbrecher,1 Anna Dolnik,1 Tamara J. Blätte,1 Dan A. Landau,4,5 Jenny Saub,1 Sven Estenfelder,1 Stefan Ibach,6 Florence Cymbalista,7 Veronique Leblond,8 Alain Delmer,9 Jasmin Bahlo,10 10 10 10 1,11 Haematologica 2020 Sandra Robrecht, Kirsten Fischer, Valentin Goede, Lars Bullinger, Volume 105(5):1379-1390 Catherine J. Wu,4 Daniel Mertens,1, Gabriella Ficz,2 John G. Gribben,2 Michael Hallek,10 Hartmut D hner1 and Stephan Stilgenbauer1 ӧ 1Department of Internal Medicine III, Ulm University, Ulm, Germany; 2Centre for Haemato-Oncology, Barts Cancer Institute, Queen Mary University of London, London, UK; 3Genomics Core Facility, Ulm University, Ulm, Germany; 4Department of Medical Oncology, Dana Farber Cancer Institute, Boston, MA, USA; 5New York Genome Center, New York, NY, USA; 6Wissenschaftlicher Service Pharma GmbH (WiSP), Langenfeld, Germany; 7Service d’Hématologie Biologique, Hôpital Avicenne, Bobigny, France; 8Service d’Hématologie, Hôpital Pitié-Salpêtrière, Paris, France; 9Service d’Hématologie Clinique, CHU de Reims, Reims, France; 10Department of Internal Medicine I, University of Cologne, Cologne, Germany and 11Department of Hematology, Oncology and Tumor Immunology, Charité University Medicine Berlin, Campus Virchow Klinikum, Berlin, Germany ABSTRACT Correspondence: o identify genomic alterations contributing to the pathogenesis of JENNIFER EDELMANN high-risk chronic lymphocytic leukemia (CLL) beyond the well- [email protected] Testablished role of TP53 aberrations, we comprehensively ana- lyzed 75 relapsed/refractory and 71 treatment-naïve high-risk cases from Received: February 5, 2019. -

Dissection of Xist Functional Elements Involved in X-Chromosome Inactivation
Dissection of Xist Functional Elements Involved in X-Chromosome Inactivation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Colognori, David A. 2019. Dissection of Xist Functional Elements Involved in X-Chromosome Inactivation. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:42029476 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Dissection of Xist functional elements involved in X-chromosome inactivation A dissertation presented by David A. Colognori to The Division of Medical Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Biological and Biomedical Sciences Harvard University Cambridge, Massachusetts May 2019 © 2019 by David A. Colognori All rights reserved. Dissertation Advisor: Jeannie T. Lee David A. Colognori Dissection of Xist functional elements involved in X-chromosome inactivation Abstract X-chromosome inactivation (XCI) is the epigenetic process of silencing one of the two X chromosomes in female mammals to balance X-linked gene dosage with that of males. This process is governed by the long noncoding RNA Xist. Xist is expressed from and coats one X chromosome in cis, leading to recruitment of various protein factors to silence gene expression. However, the functional RNA elements and molecular mechanisms involved in Xist coating and silencing remain ambiguous. -

Mouse Snw1 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Snw1 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Snw1 conditional knockout Mouse model (C57BL/6N) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Snw1 gene (NCBI Reference Sequence: NM_025507 ; Ensembl: ENSMUSG00000021039 ) is located on Mouse chromosome 12. 14 exons are identified, with the ATG start codon in exon 1 and the TAG stop codon in exon 14 (Transcript: ENSMUST00000021428). Exon 2 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Snw1 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-425D9 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 2 starts from about 0.93% of the coding region. The knockout of Exon 2 will result in frameshift of the gene. The size of intron 1 for 5'-loxP site insertion: 3484 bp, and the size of intron 2 for 3'-loxP site insertion: 3898 bp. The size of effective cKO region: ~2160 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 2 14 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Snw1 Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats.