Volibris, INN-Ambrisentan
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Endothelin System and Therapeutic Application of Endothelin Receptor
xperim ACCESS Freely available online & E en OPEN l ta a l ic P in h l a C r m f o a c l a o n l o r g u y o J Journal of ISSN: 2161-1459 Clinical & Experimental Pharmacology Research Article Endothelin System and Therapeutic Application of Endothelin Receptor Antagonists Abebe Basazn Mekuria, Zemene Demelash Kifle*, Mohammedbrhan Abdelwuhab Department of Pharmacology, School of Pharmacy, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia ABSTRACT Endothelin is a 21 amino acid molecule endogenous potent vasoconstrictor peptide. Endothelin is synthesized in vascular endothelial and smooth muscle cells, as well as in neural, renal, pulmonic, and inflammatory cells. It acts through a seven transmembrane endothelin receptor A (ETA) and endothelin receptor B (ETB) receptors belongs to G protein-coupled rhodopsin-type receptor superfamily. This peptide involved in pathogenesis of cardiovascular disorder like (heart failure, arterial hypertension, myocardial infraction and atherosclerosis), renal failure, pulmonary arterial hypertension and it also involved in pathogenesis of cancer. Potentially endothelin receptor antagonist helps the treatment of the above disorder. Currently, there are a lot of trails both per-clinical and clinical on endothelin antagonist for various cardiovascular, pulmonary and cancer disorder. Some are approved by FAD for the treatment. These agents are including both selective and non-selective endothelin receptor antagonist (ETA/B). Currently, Bosentan, Ambrisentan, and Macitentan approved -

Summary of Appeals & Independent Review Organization
All Other Appeals All other appeals are for drugs not in an inpatient hospital setting that Molina was not able to approve. Sometimes, the clinical information sent to us for these drugs do not meet medical necessity on initial review. When drug preauthorization requests are denied, a member or provider has the right to appeal. Appeals allow time to provide more clinical information. With complete clinical information, we can usually approve the drug. These are considered an appeal overturn. When the denial decision is not overturned, it is considered upheld. Service Code/Drug Name Service Code Description Number of Appeals Number of Appeals Total Appeals Upheld Overturned A9274 EXTERNAL AMB INSULIN DEL SYSTEM DISPOSABLE EA 0 1 1 Abatacept 3 1 4 Abemaciclib 1 0 1 Acalabrutinib 0 1 1 Acne Combination - Two Ingredient 1 0 1 Acyclovir 0 1 1 Adalimumab 7 14 21 Adrenergic Combination - Two Ingredient 1 0 1 Aflibercept 0 2 2 Agalsidase 1 0 1 Alfuzosin 1 0 1 Amantadine 1 0 1 Ambrisentan 0 1 1 Amphetamine 0 1 1 Amphetamine Mixtures - Two Ingredient 1 8 9 Apixaban 7 21 28 Apremilast 12 13 25 Aprepitant 0 1 1 Aripiprazole 5 9 14 ARNI-Angiotensin II Recept Antag Comb - Two Ingredient 6 9 15 Asenapine 0 1 1 Atomoxetine 1 3 4 Atorvastatin 0 1 1 Atovaquone 1 0 1 Axitinib 0 1 1 Azathioprine 0 1 1 Azilsartan 1 0 1 Azithromycin 1 0 1 Baclofen 0 1 1 Baricitinib 1 1 2 Belimumab 0 1 1 Benralizumab 1 0 1 Beta-blockers - Ophthalmic Combination - Two Ingredient 0 2 2 Bimatoprost 0 1 1 Botulinum Toxin 1 4 5 Buprenorphine 4 3 7 Calcifediol 1 0 1 Calcipotriene -

Ambrisentan (Letairis®) Issued by PHA’S Scientific Leadership Council Information Is Based on the United States Food and Drug Administration Drug Labeling
Ambrisentan (Letairis®) Issued by PHA’s Scientific Leadership Council Information is based on the United States Food and Drug Administration drug labeling Last Updated November 2013 WHAT IS AMBRISENTAN? Ambrisentan is an oral medication classified as an endothelin receptor antagonist (ERA) which is approved for the treatment of pulmonary arterial hypertension (PAH) in World Health Organization (WHO) Group 1 patients. The goal of this therapy is to improve exercise ability and slow progression of the disease. Ambrisentan was approved for PAH by the United States Food and Drug Administration (FDA) in 2007. HOW DOES AMBRISENTAN WORK? Ambrisentan works by blocking endothelin, a substance made by the body. Endothelin causes blood vessels to narrow (constrict). It also causes abnormal growth of the muscle in the walls of the blood vessels in the lungs. This narrowing increases the pressure required to push the blood through the lungs to get oxygen. By blocking the action of endothelin, causing vessels to relax, ambrisentan decreases the pulmonary blood pressure to the heart and improves its function. This generally results in the ability to be more active. Research studies have verified this improvement. HOW IS AMBRISENTAN GIVEN? Ambrisentan is taken orally, with or without food. There are two FDA approved doses; 5 mg (pale pink and square) or 10 mg (dark pink and oval). Patients’ physicians will decide which strength is right for them. HOW IS AMBRISENTAN SUPPLIED? Ambrisentan comes in 5 and 10 mg, film-coated, unscored tablets. The 5 mg tablet is pale pink and square. The 10 mg tablet is deep pink and oval. -

Ambrisentan (Letairis) Reference Number: ERX.SPMN.84 Effective Date: 07/16 Last Review Date: 06/16 Revision Log
Clinical Policy: Ambrisentan (Letairis) Reference Number: ERX.SPMN.84 Effective Date: 07/16 Last Review Date: 06/16 Revision Log See Important Reminder at the end of this policy for important regulatory and legal information. Policy/Criteria It is the policy of health plans affiliated with Envolve Pharmacy Solutions® that ambrisentan (Letairis®) is medically necessary when the following criteria are met: I. Initial Approval Criteria A. Pulmonary Arterial Hypertension (PAH) (must meet all): 1. Prescribed by or in consultation with a cardiologist or pulmonologist experienced in the diagnosis and treatment of pulmonary hypertension; 2. Diagnosis of PAH World Health Organization (WHO) Group 1 (appendix B) confirmed by right heart catheterization and with one of the following: a. Inadequate response or contraindication to acute vasodilator testing; b. Trial and failure of, or contraindication to, calcium channel blockers; 3. WHO/New York Heart Association (NYHA) functional class II, III, or IV (appendix C); 4. Prescribed dose of Letairis does not exceed 10 mg once daily. Approval Duration: 3 months B. Other diagnoses/indications: Refer to ERX.SPMN.16 - Global Biopharm Policy. II. Continued Approval A. Pulmonary Arterial Hypertension (PAH) (must meet all): 1. Currently receiving medication via health plan benefit or member has previously met all initial approval criteria. Approval Duration: 6 months B. Other diagnoses/indications (must meet 1 or 2): 1. Currently receiving medication via health plan benefit and documentation supports positive response to therapy; or 2. Refer to ERX.SPMN.16 - Global Biopharm Policy. Page 1 of 5 CLINICAL POLICY Ambrisentan Background Description/Mechanism of Action: Letairis is the brand name for ambrisentan, an endothelin receptor antagonist that is selective for the endothelin type-A (ETA) receptor. -
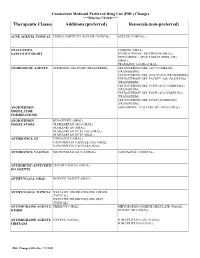
Therapeutic Classes Additions (Preferred) Removals (Non-Preferred)
Connecticut Medicaid Preferred Drug List (PDL) Changes ***Effective 7/1/2021*** Therapeutic Classes Additions (preferred) Removals (non-preferred) ACNE AGENTS, TOPICAL EPIDUO FORTE GEL W/PUMP (TOPICAL) AZELEX (TOPICAL) ANALGESICS, CODEINE (ORAL) NARCOTICS SHORT HYDROCODONE / IBUPROFEN (ORAL) OXYCODONE / APAP TABLET (PROLATE) (ORAL) TRAMADOL 100 MG (ORAL) ANDROGENIC AGENTS ANDROGEL GEL PUMP (TRANSDERM) TESTOSTERONE GEL (AG) (VOGELXO) (TRANSDERM) TESTOSTERONE GEL (VOGELXO) (TRANSDERM) TESTOSTERONE GEL PACKET (AG) (VOGELXO) (TRANSDERM) TESTOSTERONE GEL PUMP (AG) (ANDROGEL) (TRANSDERM) TESTOSTERONE GEL PUMP (AG) (VOGELXO) (TRANSDERM) TESTOSTERONE GEL PUMP (ANDROGEL) (TRANSDERM) ANGIOTENSIN AMLODIPINE / VALSARTAN / HCTZ (ORAL) MODULATOR COMBINATIONS ANGIOTENSIN BENAZEPRIL (ORAL) MODULATORS OLMESARTAN (AG) (ORAL) OLMESARTAN (ORAL) OLMESARTAN HCTZ (AG) (ORAL) OLMESARTAN HCTZ (ORAL) ANTIBIOTICS, GI TINIDAZOLE (ORAL) VANCOMYCIN CAPSULE (AG) (ORAL) VANCOMYCIN CAPSULE (ORAL) ANTIBIOTICS, VAGINAL METRONIDAZOLE (VAGINAL) VANDAZOLE (VAGINAL) ANTIEMETIC/ANTIVERTI EMEND CAPSULE (ORAL) GO AGENTS ANTIFUNGALS, ORAL NOXAFIL TABLET (ORAL) ANTIFUNGALS, TOPICAL NYSTATIN-TRIAMCINOLONE CREAM (TOPICAL) NYSTATIN-TRIAMCINOLONE OINT (TOPICAL) ANTIMIGRAINE AGENTS, UBRELVY (ORAL) DIHYDROERGOTAMINE MESYLATE (NASAL) OTHER NURTEC ODT (ORAL) ANTIMIGRAINE AGENTS, IMITREX (NASAL) SUMATRIPTAN (AG) (NASAL) TRIPTANS SUMATRIPTAN (NASAL) PDL Changes Effective: 7/1/2021 Connecticut Medicaid Preferred Drug List (PDL) Changes ***Effective 7/1/2021*** Therapeutic Classes Additions -

84 November 2008 Produced by NHS Tayside Drug and Therapeutics Committee Medicines Advisory Group (MAG)
TAYSIDE PRESCRIBER Tayside DTC Supplement No 84 November 2008 Produced by NHS Tayside Drug and Therapeutics Committee Medicines Advisory Group (MAG) SMC Advice issued in November 2008 Medicine Indication Local recommendation Comments and useful category links Ambrisentan tablets Treatment of patients with class Restricted for use within SMC advice (Volibris®) II and III pulmonary arterial national treatment centre SPC link hypertension to improve exercise capacity Anidulafungin powder and Treatment of invasive Pending AMG decision SMC advice solvent (Ecalta®) - candidiasis in adult non- SPC link Resubmission neutropenic patients Bosentan film-coated tablets Class II pulmonary arterial Not recommended SMC advice (Tracleer®) - hypertension Bosentan was previously Non submission accepted in 2003 for restricted use in grade III pulmonary arterial hypertension Icatibant solution for Symptomatic treatment of acute Not recommended SMC advice subcutaneous injection attacks of hereditary (Firazyr®) angioedema in adults Insulin glulisine solution for Treatment of adolescents and Non-formulary SMC advice subcutaneous injection children, 6 years or older with SPC link (Apidra®) - Abbreviated diabetes mellitus where a short Diabetes Handbook submission acting insulin analogue is appropriate Insulin lispro solution for Adults and children with Non-formulary SMC advice injection (Humalog® diabetes mellitus SPC link KwikPen) - Diabetes Handbook Abbreviated submission Insulin lispro (Humalog® Patients with diabetes mellitus, Non-formulary SMC advice Mix25 -

List of Formulary Drug Removals
July 2021 Formulary Drug Removals Below is a list of medicines by drug class that have been removed from your plan’s formulary. If you continue using one of the drugs listed below and identified as a Formulary Drug Removal, you may be required to pay the full cost. If you are currently using one of the formulary drug removals, ask your doctor to choose one of the generic or brand formulary options listed below. Category Formulary Drug Formulary Options Drug Class Removals Acromegaly SANDOSTATIN LAR SOMATULINE DEPOT SIGNIFOR LAR SOMAVERT Allergies dexchlorpheniramine levocetirizine Antihistamines Diphen Elixir RyClora CARBINOXAMINE TABLET 6 MG Allergies BECONASE AQ flunisolide spray, fluticasone spray, mometasone spray, DYMISTA Nasal Steroids / Combinations OMNARIS QNASL ZETONNA Anticonvulsants topiramate ext-rel capsule carbamazepine, carbamazepine ext-rel, clobazam, divalproex sodium, (generics for QUDEXY XR only) divalproex sodium ext-rel, gabapentin, lamotrigine, lamotrigine ext-rel, levetiracetam, levetiracetam ext-rel, oxcarbazepine, phenobarbital, phenytoin, phenytoin sodium extended, primidone, rufinamide, tiagabine, topiramate, valproic acid, zonisamide, FYCOMPA, OXTELLAR XR, TROKENDI XR, VIMPAT, XCOPRI BANZEL SUSPENSION clobazam, lamotrigine, rufinamide, topiramate, TROKENDI XR ONFI SABRIL vigabatrin ZONEGRAN carbamazepine, carbamazepine ext-rel, divalproex sodium, divalproex sodium ext-rel, gabapentin, lamotrigine, lamotrigine ext-rel, levetiracetam, levetiracetam ext-rel, oxcarbazepine, phenobarbital, phenytoin, phenytoin sodium -

Pennsylvania Department of Human Services Statewide Preferred Drug List (PDL)*
Pennsylvania Department of Human Services Statewide Preferred Drug List (PDL)* *The Statewide PDL is not an all-inclusive list of drugs covered by Medical Assistance. Drugs in Statewide PDL classes that are new to market will be non-preferred until reviewed by the DHS Pharmacy and Therapeutics Committee. Fee-for-Service‡ (FFS) Pharmacy General Prior Authorization Requirements: https://dhs.pa.gov/providers/Pharmacy-Services/Pages/Pharmacy-Prior-Authorization-General-Requirements.aspx FFS† Pharmacy Prior Authorization Clinical Guidelines: https://www.dhs.pa.gov/providers/Pharmacy-Services/Pages/Clinical-Guidelines.aspx FFS‡ Pharmacy Prior Authorization Fax Forms: https://www.dhs.pa.gov/providers/Pharmacy-Services/Pages/Pharmacy-Services-Fax-Forms.aspx FFS‡ Pharmacy Quantity Limits/Daily Dose Limits: https://dhs.pa.gov/providers/Pharmacy-Services/Pages/Quantity-Limits-and-Daily-Dose-Limits.aspx ‡This information is specific to FFS. Please refer to each managed care organization’s (MCO) website for MCO prior authorization procedures, prior authorization fax request forms, and quantity limits. †Prior authorization guidelines for drugs and products included in the Statewide PDL apply to FFS and the Pennsylvania Medical Assistance MCOs. Prior authorization guidelines for drugs and products not included in the Statewide PDL are specific to FFS. Please refer to each MCO’s website for MCO-specific prior authorization requirements for drugs and products not included in the Statewide PDL. ACNE AGENTS, ORAL Preferred Agents Non-Preferred Agents Amnesteem -

Ambrisentan (Letairis and Opsumit)
US Family Health Plan Prior Authorization Request Form for ambrisentan (Letairis), macitentan (Opsumit) To be completed and signed by the prescriber. To be used only for prescriptions which are to be filled through the Department of Defense (DoD) US Family Health Plan Pharmacy Program. US Family Health Plan is a TRICARE contractor for DoD. The completed form may be faxed to 855-273-5735 OR The patient may attach the completed form to the prescription and mail it to: Attn: Pharmacy, 77 Warren St, Brighton, MA 02135 QUESTIONS? Call 1-877-880-7007 1B Step 3BPlease complete patient and physician information (please print): 2B1 Patient Name: Physician Name: Address: Address: Sponsor ID # Phone #: Date of Birth: Secure Fax #: 4B Step 6BPlease complete the clinical assessment: 5B2 1. 7BDoes the patient have a documented diagnosis of WHO Yes No group 1 PAH? Proceed to question 2 STOP Cov erage not approv ed 2. 8BIs the requested medication being prescribed by or in Yes No consultation with a cardiologist or a pulmonologist? Proceed to question 3 STOP Cov erage not approv ed 3. 9BHas the patient had a right heart catheterization? Yes No Proceed to question 4 STOP Cov erage not approv ed 4. 10BIs documentation being provided to confirm that the patient Yes No has had a right heart catheterization? Proceed to question 5 STOP PLEASE NOTE: Medical documentation specific to your Cov erage not approv ed response to this question must be attached to this case or your request could be denied. Documentation may include, but is not limited to, chart notes and catheterization laboratory reports. -

Wednesday, April 8, 2015 4 P.M
Wednesday, April 8, 2015 4 p.m. Oklahoma Health Care Authority 4345 N. Lincoln Blvd. Oklahoma City, OK 73105 The University of Oklahoma Health Sciences Center COLLEGE OF PHARMACY PHARMACY MANAGEMENT CONSULTANTS MEMORANDUM TO: Drug Utilization Review Board Members FROM: Bethany Holderread, Pharm.D. SUBJECT: Packet Contents for Board Meeting – April 8, 2015 DATE: April 1, 2015 NOTE: The DUR Board will meet at 4:00 p.m. The meeting will be held at 4345 N Lincoln Blvd. Enclosed are the following items related to the April meeting. Material is arranged in order of the agenda. Call to Order Public Comment Forum Action Item – Approval of DUR Board Meeting Minutes – See Appendix A Update on Medication Coverage Authorization Unit/Oral Viscous Lidocaine Claims Analysis Update – Appendix B Fiscal Year 2014 Annual Review of SoonerCare Pharmacy Benefit – Appendix C Action Item – Vote to Prior Authorize Sylvant™ (Siltuximab) – Appendix D Action Item – Vote to Prior Authorize Ecoza™ (Econazole Nitrate), Jublia® (Efinaconazole), and Kerydin™ (Tavaborole) – Appendix E Action Item – Vote to Prior Authorize Izba® (Travoprost Ophthalmic Solution) – Appendix F Annual Review of Antihypertensive Medications and 30-Day Notice to Prior Authorize Hemangeol™ (Propranolol Oral Solution), Sotylize™ (Sotalol Oral Solution), and Prestalia® (Perindopril/Amlodipine) – Appendix G Annual Review of Hereditary Angioedema Medications and 30-Day Notice to Prior Authorize Ruconest® (C1 Esterase Inhibitor) – Appendix H Annual Review of Diabetes Medications and 30-Day Notice to Prior Authorize Tanzeum™ (Albiglutide), Trulicity™ (Dulaglutide), Cycloset® (Bromocriptine), Jardiance® (Empagliflozin), Invokamet™ (Canagliflozin/Metformin), Xigduo™ XR (Dapagliflozin/Metformin Extended-Release), Glyxambi® (Empagliflozin/Linagliptin), Afrezza® (Insulin Human Inhalation Powder), and Toujeo® (Insulin Glargine) – Appendix I Annual Review of Pediculicides – Appendix J FDA and DEA Updates – Appendix K Future Business Adjournment ORI-4403 • P.O. -
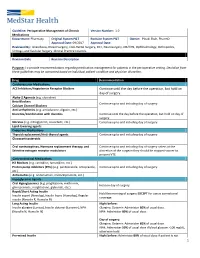
1 Continue Until the Day Before the Operation, but Hold on Day of Surgery
Guideline: Perioperative Management of Chronic Version Number: 1.0 Medications Department: Pharmacy Original System P&T Revision System P&T Owner: Pinaki Shah, PharmD Approval Date: 09/2017 Approval Date: Reviewed By: Anesthesia, Breast Surgery, Colo-Rectal Surgery, ENT, Neurosurgery, OB/GYN, Ophthalmology, Orthopedics, Urology, and Vascular Surgery Clinical Practice Councils. Revision Date Revision Description Purpose: To provide recommendations regarding medication management for patients in the perioperative setting. Deviation from these guidelines may be warranted based on individual patient condition and physician discretion. Drug Recommendation Cardiovascular Medications ACE Inhibitors/Angiotensin Receptor Blockers Continue until the day before the operation, but hold on day of surgery. Alpha-2 Agonists (e.g. clonidine) Beta Blockers Continue up to and including day of surgery. Calcium Channel Blockers Anti-arrhythmics (e.g. amiodarone, digoxin, etc.) Diuretics/Combination with diuretics Continue until the day before the operation, but hold on day of surgery. Nitrates (e.g. nitroglycerin, isosorbide, etc.) Continue up to and including day of surgery. Lipid-lowering agents Endocrine Medications Thyroid replacement/Anti-thyroid agents Continue up to and including day of surgery. Glucocorticosteroids Oral contraceptives, Hormone replacement therapy, and Continue up to and including day of surgery unless at the Selective estrogen receptor modulators discretion of the surgeon they should be stopped sooner to prevent VTE. Gastrointestinal Medications H2 Blockers (e.g. ranitidine, famotidine, etc.) Proton pump inhibitors (PPIs) (e.g. pantoprazole, omeprazole, Continue up to and including day of surgery. etc.) Antiemetics (e.g. ondansetron, metoclopramide, etc.) Hypoglycemic Agents Oral Hypoglycemics (e.g. pioglitazone, metformin, Hold on day of surgery. glimepramide, resiglitazone, glyburide, etc.) Rapid/Short Acting Insulin Hold the morning of surgery EXCEPT for use as correctional Insulin aspart (Novolog), Insulin lispro (Humalog), Regular coverage. -

Endothelin ETA Receptor Blockade, by Activating ETB Receptors, Increases Vascular Permeability and Induces Exaggerated Fluid Retention S
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2017/02/21/jpet.116.234930.DC1 1521-0103/361/2/322–333$25.00 https://doi.org/10.1124/jpet.116.234930 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 361:322–333, May 2017 Copyright ª 2017 by The American Society for Pharmacology and Experimental Therapeutics Endothelin ETA Receptor Blockade, by Activating ETB Receptors, Increases Vascular Permeability and Induces Exaggerated Fluid Retention s Magali Vercauteren, Frederic Trensz, Anne Pasquali, Christophe Cattaneo, Daniel S. Strasser, Patrick Hess, Marc Iglarz, and Martine Clozel Drug Discovery Department, Actelion Pharmaceuticals Ltd., Allschwil, Switzerland Received May 9, 2016; accepted February 17, 2017 Downloaded from ABSTRACT Endothelin (ET) receptor antagonists have been associated with ETA-selective antagonism increased vascular permeability via ETB fluid retention. It has been suggested that, of the two endothelin receptor overstimulation. Acutely, ETA-selective but not dual receptor subtypes, ETB receptors should not be blocked, because antagonism activated sympathetic activity and increased plasma of their involvement in natriuresis and diuresis. Surprisingly, arginine vasopressin and aldosterone concentrations. The hem- clinical data suggest that ETA-selective antagonists pose a greater atocrit/hemoglobin decrease induced by ETA-selective antago- jpet.aspetjournals.org risk of fluid overload than dual antagonists. The purpose of this nism was reduced in Brattleboro rats and in Wistar rats treated study was to evaluate the contribution of each endothelin receptor with an arginine vasopressin receptor antagonist. Finally, the to fluid retention and vascular permeability in rats. Sitaxentan and decrease in hematocrit/hemoglobin was larger in the venous than ambrisentan as ETA-selective antagonists and bosentan and in the arterial side, suggesting fluid redistribution.