The Control of Inflammation in Airway Epithelial Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reprogramming of Trna Modifications Controls the Oxidative Stress Response by Codon-Biased Translation of Proteins
Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Chan, Clement T.Y. et al. “Reprogramming of tRNA Modifications Controls the Oxidative Stress Response by Codon-biased Translation of Proteins.” Nature Communications 3 (2012): 937. As Published http://dx.doi.org/10.1038/ncomms1938 Publisher Nature Publishing Group Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/76775 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins Clement T.Y. Chan,1,2 Yan Ling Joy Pang,1 Wenjun Deng,1 I. Ramesh Babu,1 Madhu Dyavaiah,3 Thomas J. Begley3 and Peter C. Dedon1,4* 1Department of Biological Engineering, 2Department of Chemistry and 4Center for Environmental Health Sciences, Massachusetts Institute of Technology, Cambridge, MA 02139; 3College of Nanoscale Science and Engineering, University at Albany, SUNY, Albany, NY 12203 * Corresponding author: PCD, Department of Biological Engineering, NE47-277, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139; tel 617-253-8017; fax 617-324-7554; email [email protected] 2 ABSTRACT Selective translation of survival proteins is an important facet of cellular stress response. We recently demonstrated that this translational control involves a stress-specific reprogramming of modified ribonucleosides in tRNA. -

Allele-Specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish
Allele-specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish by Ailu Chen A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 1, 2015 Keywords: catfish, interspecific hybrids, allele-specific expression, ribosomal protein Copyright 2015 by Ailu Chen Approved by Zhanjiang Liu, Chair, Professor, School of Fisheries, Aquaculture and Aquatic Sciences Nannan Liu, Professor, Entomology and Plant Pathology Eric Peatman, Associate Professor, School of Fisheries, Aquaculture and Aquatic Sciences Aaron M. Rashotte, Associate Professor, Biological Sciences Abstract Interspecific hybridization results in a vast reservoir of allelic variations, which may potentially contribute to phenotypical enhancement in the hybrids. Whether the allelic variations are related to the downstream phenotypic differences of interspecific hybrid is still an open question. The recently developed genome-wide allele-specific approaches that harness high- throughput sequencing technology allow direct quantification of allelic variations and gene expression patterns. In this work, I investigated allele-specific expression (ASE) pattern using RNA-Seq datasets generated from interspecific catfish hybrids. The objective of the study is to determine the ASE genes and pathways in which they are involved. Specifically, my study investigated ASE-SNPs, ASE-genes, parent-of-origins of ASE allele and how ASE would possibly contribute to heterosis. My data showed that ASE was operating in the interspecific catfish system. Of the 66,251 and 177,841 SNPs identified from the datasets of the liver and gill, 5,420 (8.2%) and 13,390 (7.5%) SNPs were identified as significant ASE-SNPs, respectively. -

Propranolol-Mediated Attenuation of MMP-9 Excretion in Infants with Hemangiomas
Supplementary Online Content Thaivalappil S, Bauman N, Saieg A, Movius E, Brown KJ, Preciado D. Propranolol-mediated attenuation of MMP-9 excretion in infants with hemangiomas. JAMA Otolaryngol Head Neck Surg. doi:10.1001/jamaoto.2013.4773 eTable. List of All of the Proteins Identified by Proteomics This supplementary material has been provided by the authors to give readers additional information about their work. © 2013 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/01/2021 eTable. List of All of the Proteins Identified by Proteomics Protein Name Prop 12 mo/4 Pred 12 mo/4 Δ Prop to Pred mo mo Myeloperoxidase OS=Homo sapiens GN=MPO 26.00 143.00 ‐117.00 Lactotransferrin OS=Homo sapiens GN=LTF 114.00 205.50 ‐91.50 Matrix metalloproteinase‐9 OS=Homo sapiens GN=MMP9 5.00 36.00 ‐31.00 Neutrophil elastase OS=Homo sapiens GN=ELANE 24.00 48.00 ‐24.00 Bleomycin hydrolase OS=Homo sapiens GN=BLMH 3.00 25.00 ‐22.00 CAP7_HUMAN Azurocidin OS=Homo sapiens GN=AZU1 PE=1 SV=3 4.00 26.00 ‐22.00 S10A8_HUMAN Protein S100‐A8 OS=Homo sapiens GN=S100A8 PE=1 14.67 30.50 ‐15.83 SV=1 IL1F9_HUMAN Interleukin‐1 family member 9 OS=Homo sapiens 1.00 15.00 ‐14.00 GN=IL1F9 PE=1 SV=1 MUC5B_HUMAN Mucin‐5B OS=Homo sapiens GN=MUC5B PE=1 SV=3 2.00 14.00 ‐12.00 MUC4_HUMAN Mucin‐4 OS=Homo sapiens GN=MUC4 PE=1 SV=3 1.00 12.00 ‐11.00 HRG_HUMAN Histidine‐rich glycoprotein OS=Homo sapiens GN=HRG 1.00 12.00 ‐11.00 PE=1 SV=1 TKT_HUMAN Transketolase OS=Homo sapiens GN=TKT PE=1 SV=3 17.00 28.00 ‐11.00 CATG_HUMAN Cathepsin G OS=Homo -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Protein Identities in Evs Isolated from U87-MG GBM Cells As Determined by NG LC-MS/MS
Protein identities in EVs isolated from U87-MG GBM cells as determined by NG LC-MS/MS. No. Accession Description Σ Coverage Σ# Proteins Σ# Unique Peptides Σ# Peptides Σ# PSMs # AAs MW [kDa] calc. pI 1 A8MS94 Putative golgin subfamily A member 2-like protein 5 OS=Homo sapiens PE=5 SV=2 - [GG2L5_HUMAN] 100 1 1 7 88 110 12,03704523 5,681152344 2 P60660 Myosin light polypeptide 6 OS=Homo sapiens GN=MYL6 PE=1 SV=2 - [MYL6_HUMAN] 100 3 5 17 173 151 16,91913397 4,652832031 3 Q6ZYL4 General transcription factor IIH subunit 5 OS=Homo sapiens GN=GTF2H5 PE=1 SV=1 - [TF2H5_HUMAN] 98,59 1 1 4 13 71 8,048185945 4,652832031 4 P60709 Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 - [ACTB_HUMAN] 97,6 5 5 35 917 375 41,70973209 5,478027344 5 P13489 Ribonuclease inhibitor OS=Homo sapiens GN=RNH1 PE=1 SV=2 - [RINI_HUMAN] 96,75 1 12 37 173 461 49,94108966 4,817871094 6 P09382 Galectin-1 OS=Homo sapiens GN=LGALS1 PE=1 SV=2 - [LEG1_HUMAN] 96,3 1 7 14 283 135 14,70620005 5,503417969 7 P60174 Triosephosphate isomerase OS=Homo sapiens GN=TPI1 PE=1 SV=3 - [TPIS_HUMAN] 95,1 3 16 25 375 286 30,77169764 5,922363281 8 P04406 Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 - [G3P_HUMAN] 94,63 2 13 31 509 335 36,03039959 8,455566406 9 Q15185 Prostaglandin E synthase 3 OS=Homo sapiens GN=PTGES3 PE=1 SV=1 - [TEBP_HUMAN] 93,13 1 5 12 74 160 18,68541938 4,538574219 10 P09417 Dihydropteridine reductase OS=Homo sapiens GN=QDPR PE=1 SV=2 - [DHPR_HUMAN] 93,03 1 1 17 69 244 25,77302971 7,371582031 11 P01911 HLA class II histocompatibility antigen, -

Α Are Regulated by Heat Shock Protein 90
The Levels of Retinoic Acid-Inducible Gene I Are Regulated by Heat Shock Protein 90- α Tomoh Matsumiya, Tadaatsu Imaizumi, Hidemi Yoshida, Kei Satoh, Matthew K. Topham and Diana M. Stafforini This information is current as of October 2, 2021. J Immunol 2009; 182:2717-2725; ; doi: 10.4049/jimmunol.0802933 http://www.jimmunol.org/content/182/5/2717 Downloaded from References This article cites 44 articles, 19 of which you can access for free at: http://www.jimmunol.org/content/182/5/2717.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on October 2, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2009 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology The Levels of Retinoic Acid-Inducible Gene I Are Regulated by Heat Shock Protein 90-␣1 Tomoh Matsumiya,*‡ Tadaatsu Imaizumi,‡ Hidemi Yoshida,‡ Kei Satoh,‡ Matthew K. Topham,*† and Diana M. Stafforini2*† Retinoic acid-inducible gene I (RIG-I) is an intracellular pattern recognition receptor that plays important roles during innate immune responses to viral dsRNAs. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

(12) United States Patent (10) Patent No.: US 8,609,416 B2 Barnett (45) Date of Patent: Dec
USOO8609416B2 (12) United States Patent (10) Patent No.: US 8,609,416 B2 Barnett (45) Date of Patent: Dec. 17, 2013 (54) METHODS AND COMPOSITIONS OTHER PUBLICATIONS COMPRISING HEAT SHOCKPROTEINS Novoselova et al., “Treatment with extracellular HSP70/HSC70 pro (75) Inventor: Michael E. Barnett, Manhattan, KS tein can reduce polyglutamine toxicity and aggregation.” J (US) Neurochem 94:597-606, 2005.* Johnson et al., (1993) Exogenous HSP70 becomes cell associated but (73) Assignee: Ventria Bioscience, Fort Collins, CO not internalized, by stressed arterial Smooth muscle cell. In vitro (US) Cellular and Developmental Biology—Animal, vol. 29A. No. 10, pp. 807-821. (*) Notice: Subject to any disclaimer, the term of this Bethke et al., (2002) Different efficiency of heat shock proteins patent is extended or adjusted under 35 (HSP) to activate human monocytes and dendritic cells; Superiority of U.S.C. 154(b) by 244 days. HSP60, The Journal of Immunology, vol. 169, pp. 6141-6148. Khan et al., (2008) Toll-like receptor 4-mediated growth of (21) Appl. No.: 12/972,112 endometriosis by human heat-shock protein 70, Human Reproduc tion, vol. 23, No. 10, pp. 2210-2219. (22) Filed: Dec. 17, 2010 Lasunskaia E.B., et al., (2003) Transfection of NSO myeloma fusion partner cells with HSP70 gene results in higher hybridoma yield by (65) Prior Publication Data improving cellular resistance to apoptosis, Biotechnology and Bioengineering 81 (4):496-504. US 2011 FO189751A1 Aug. 4, 2011 * cited by examiner Related U.S. Application Data (60) Provisional application No. 61/288,234, filed on Dec. Primary Examiner — Rosanne Kosson 18, 2009. -

The Plasma Peptides of Alzheimer's Disease
Florentinus‑Mefailoski et al. Clin Proteom (2021) 18:17 https://doi.org/10.1186/s12014‑021‑09320‑2 Clinical Proteomics RESEARCH Open Access The plasma peptides of Alzheimer’s disease Angelique Florentinus‑Mefailoski1, Peter Bowden1, Philip Scheltens2, Joep Killestein3, Charlotte Teunissen4 and John G. Marshall1,5* Abstract Background: A practical strategy to discover proteins specifc to Alzheimer’s dementia (AD) may be to compare the plasma peptides and proteins from patients with dementia to normal controls and patients with neurological condi‑ tions like multiple sclerosis or other diseases. The aim was a proof of principle for a method to discover proteins and/ or peptides of plasma that show greater observation frequency and/or precursor intensity in AD. The endogenous tryptic peptides of Alzheimer’s were compared to normals, multiple sclerosis, ovarian cancer, breast cancer, female normal, sepsis, ICU Control, heart attack, along with their institution‑matched controls, and normal samples collected directly onto ice. Methods: Endogenous tryptic peptides were extracted from blinded, individual AD and control EDTA plasma sam‑ ples in a step gradient of acetonitrile for random and independent sampling by LC–ESI–MS/MS with a set of robust and sensitive linear quadrupole ion traps. The MS/MS spectra were ft to fully tryptic peptides within proteins identi‑ fed using the X!TANDEM algorithm. Observation frequency of the identifed proteins was counted using SEQUEST algorithm. The proteins with apparently increased observation frequency in AD versus AD Control were revealed graphically and subsequently tested by Chi Square analysis. The proteins specifc to AD plasma by Chi Square with FDR correction were analyzed by the STRING algorithm. -

Cloud-Clone 16-17
Cloud-Clone - 2016-17 Catalog Description Pack Size Supplier Rupee(RS) ACB028Hu CLIA Kit for Anti-Albumin Antibody (AAA) 96T Cloud-Clone 74750 AEA044Hu ELISA Kit for Anti-Growth Hormone Antibody (Anti-GHAb) 96T Cloud-Clone 74750 AEA255Hu ELISA Kit for Anti-Apolipoprotein Antibodies (AAHA) 96T Cloud-Clone 74750 AEA417Hu ELISA Kit for Anti-Proteolipid Protein 1, Myelin Antibody (Anti-PLP1) 96T Cloud-Clone 74750 AEA421Hu ELISA Kit for Anti-Myelin Oligodendrocyte Glycoprotein Antibody (Anti- 96T Cloud-Clone 74750 MOG) AEA465Hu ELISA Kit for Anti-Sperm Antibody (AsAb) 96T Cloud-Clone 74750 AEA539Hu ELISA Kit for Anti-Myelin Basic Protein Antibody (Anti-MBP) 96T Cloud-Clone 71250 AEA546Hu ELISA Kit for Anti-IgA Antibody 96T Cloud-Clone 71250 AEA601Hu ELISA Kit for Anti-Myeloperoxidase Antibody (Anti-MPO) 96T Cloud-Clone 71250 AEA747Hu ELISA Kit for Anti-Complement 1q Antibody (Anti-C1q) 96T Cloud-Clone 74750 AEA821Hu ELISA Kit for Anti-C Reactive Protein Antibody (Anti-CRP) 96T Cloud-Clone 74750 AEA895Hu ELISA Kit for Anti-Insulin Receptor Antibody (AIRA) 96T Cloud-Clone 74750 AEB028Hu ELISA Kit for Anti-Albumin Antibody (AAA) 96T Cloud-Clone 71250 AEB264Hu ELISA Kit for Insulin Autoantibody (IAA) 96T Cloud-Clone 74750 AEB480Hu ELISA Kit for Anti-Mannose Binding Lectin Antibody (Anti-MBL) 96T Cloud-Clone 88575 AED245Hu ELISA Kit for Anti-Glutamic Acid Decarboxylase Antibodies (Anti-GAD) 96T Cloud-Clone 71250 AEK505Hu ELISA Kit for Anti-Heparin/Platelet Factor 4 Antibodies (Anti-HPF4) 96T Cloud-Clone 71250 CCA005Hu CLIA Kit for Angiotensin II -

Molecular Cloning and Characterization of Cdna Encoding a Putative Stress-Induced Heat-Shock Protein from Camelus Dromedarius
Int. J. Mol. Sci. 2011, 12, 4214-4236; doi:10.3390/ijms12074214 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article Molecular Cloning and Characterization of cDNA Encoding a Putative Stress-Induced Heat-Shock Protein from Camelus dromedarius Mohamed S. Elrobh *, Mohammad S. Alanazi, Wajahatullah Khan, Zainularifeen Abduljaleel, Abdullah Al-Amri and Mohammad D. Bazzi Genomic Research Chair Unit, Department of Biochemistry, College of Science, King Saud University, PO Box 2455, Riyadh 11451, Saudi Arabia; E-Mails: [email protected] (M.S.A.); [email protected] (W.K.); [email protected] (Z.A.); [email protected] (A.A.-A.) [email protected] (M.D.B.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +966-146-759-44; Fax: +966-146-757-91. Received: 5 May 2011; in revised form: 9 June 2011; / Accepted: 15 June 2011 / Published: 27 June 2011 Abstract: Heat shock proteins are ubiquitous, induced under a number of environmental and metabolic stresses, with highly conserved DNA sequences among mammalian species. Camelus dromedaries (the Arabian camel) domesticated under semi-desert environments, is well adapted to tolerate and survive against severe drought and high temperatures for extended periods. This is the first report of molecular cloning and characterization of full length cDNA of encoding a putative stress-induced heat shock HSPA6 protein (also called HSP70B′) from Arabian camel. A full-length cDNA (2417 bp) was obtained by rapid amplification of cDNA ends (RACE) and cloned in pET-b expression vector. The sequence analysis of HSPA6 gene showed 1932 bp-long open reading frame encoding 643 amino acids. -
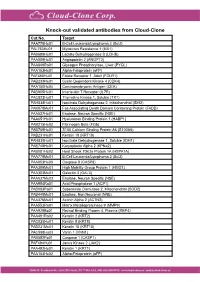
Knock-Out Validated Antibodies from Cloud-Clone Cat.No
Knock-out validated antibodies from Cloud-Clone Cat.No. Target PAA778Hu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAL763Hu01 Myxovirus Resistance 1 (MX1) PAB698Hu01 Lactate Dehydrogenase B (LDHB) PAA009Hu01 Angiopoietin 2 (ANGPT2) PAA849Ra01 Glycogen Phosphorylase, Liver (PYGL) PAA153Hu01 Alpha-Fetoprotein (aFP) PAF460Hu01 Folate Receptor 1, Adult (FOLR1) PAB233Hu01 Cyclin Dependent Kinase 4 (CDK4) PAA150Hu04 Carcinoembryonic Antigen (CEA) PAB905Hu01 Interleukin 7 Receptor (IL7R) PAC823Hu01 Thymidine Kinase 1, Soluble (TK1) PAH838Hu01 Isocitrate Dehydrogenase 2, mitochondrial (IDH2) PAK078Mu01 Fas Associating Death Domain Containing Protein (FADD) PAA537Hu01 Enolase, Neuron Specific (NSE) PAA651Hu01 Hyaluronan Binding Protein 1 (HABP1) PAB215Hu02 Fibrinogen Beta (FGb) PAB769Hu01 S100 Calcium Binding Protein A6 (S100A6) PAB231Hu01 Keratin 18 (KRT18) PAH839Hu01 Isocitrate Dehydrogenase 1, Soluble (IDH1) PAE748Hu01 Karyopherin Alpha 2 (KPNa2) PAB081Hu02 Heat Shock 70kDa Protein 1A (HSPA1A) PAA778Mu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAA853Hu03 Caspase 8 (CASP8) PAA399Mu01 High Mobility Group Protein 1 (HMG1) PAA303Mu01 Galectin 3 (GAL3) PAA537Mu02 Enolase, Neuron Specific (NSE) PAA994Ra01 Acid Phosphatase 1 (ACP1) PAB083Ra01 Superoxide Dismutase 2, Mitochondrial (SOD2) PAB449Mu01 Enolase, Non Neuronal (NNE) PAA376Mu01 Actinin Alpha 2 (ACTN2) PAA553Ra01 Matrix Metalloproteinase 9 (MMP9) PAA929Bo01 Retinol Binding Protein 4, Plasma (RBP4) PAA491Ra02 Keratin 2 (KRT2) PAC025Hu01 Keratin 8 (KRT8) PAB231Mu01 Keratin 18 (KRT18) PAC598Hu03 Vanin 1 (VNN1)